
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFundamental aspects of property tuning in push–pull molecules†
Filip
Bureš
*
Institute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, Pardubice, Czech Republic. E-mail: filip.bures@upce.cz; Web: http://bures.upce.cz Fax: +420 46 603 7068; Tel: +420 46 603 7099
First published on 22nd October 2014
Abstract
Property tuning in selected examples of D–π–A molecules has been discussed and summarized in this review article. The tuning and structure–property relationships have been demonstrated on the particular A, π and D parts of the push–pull molecule. Special emphasis has been put on the tuning of the FMO levels and optical properties. Further prospective applications of the given chromophore have also been considered.
1. Introduction
Nowadays, organic molecules with a delocalized π-system of electrons represent attractive targets for application in advanced functional materials. Due to their easy synthesis, well-defined and tuneable structure, molecular arrangement, and predictable, peculiar and unique properties, (hetero)aromatic π-conjugated systems are a notable feature of materials science. Within the last four or five decades, organic π-systems have become an extensively investigated, burgeoning area of organic chemistry, and have found widespread applications across various branches of modern research. They can be found as active substances in organic electronics and optoelectronics,1 conductors,2 photovoltaics3 and solar photon conversion.4 Based on the development in these fields, and in parallel to widely explored inorganic analogues, organic devices such as field-effect transistors (OFET),5 light-emitting diodes (OLED),6 photovoltaic cells (OPVC),7 dye-sensitizing solar cells (DSSC)8 and bulk-heterojunction solar cells (BHJ)9 belong to well established and known applications of π-conjugated molecules.Organic π-systems end-capped with an electron donor (D) and an electron acceptor (A) represent a subclass of the aforementioned molecules widely known as push–pull systems. D–A interaction, or so-called intramolecular charge-transfer (ICT), in such D–π–A molecules accounts for even more distinct optoelectronic properties. Due to the D–A interaction, a new low-energy molecular orbital (MO) is being formed. Facile excitation of the electrons within the new MO can be achieved using visible light, and therefore push–pull molecules are generally coloured and referred to as charge-transfer chromophores.10 The ICT is also responsible for the polarization of the push–pull chromophore and generation of a molecular dipole. The extent of the ICT can be expressed by two limiting resonance forms, as shown in Fig. 1 for a simple D–π–A model based on nitroaniline. The impact of the ICT on the optical properties of π-conjugated molecules can be demonstrated by the UV/Vis absorption spectra measured for aniline, nitrobenzene, 4-nitroaniline and 3-nitroaniline (Fig. 1). Whereas for the first two compounds only absorption in the UV region is being observed, 4-nitroaniline showed intense and bathochromically shifted longest-wavelength absorption maxima (CT-band). On the contrary, 3-nitroaniline showed only a diminished CT-band as a result of a non-conjugating arrangement of the amino donor and the nitro acceptor.
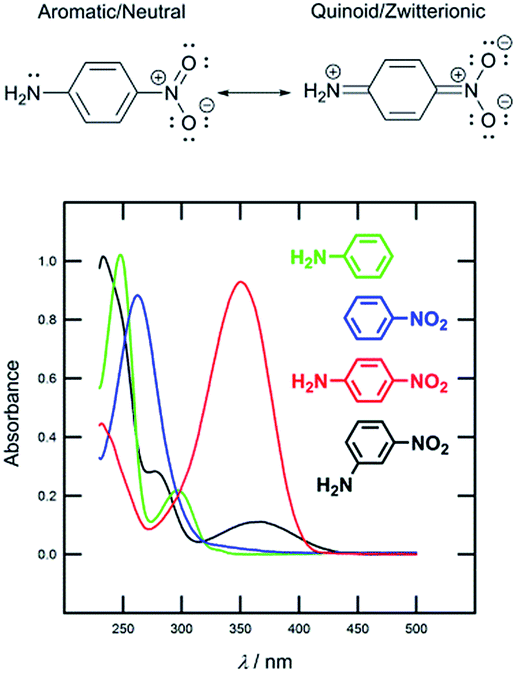 | ||
| Fig. 1 Limiting resonance forms and UV/Vis spectra for model D–π–A systems based on nitroaniline. | ||
Typical electron donors D are represented by the substituents with +M/+I effects such as OH, NH2, OR and NR2, heterocyclic moieties such as thiophene11 and proaromatic pyran-4-ylidines12 as well as some metallocenes.13,14 On the other hand, the most used electron acceptors A involve substituents featuring −M/−I effects such as NO2, CN, CHO and electron deficient heterocyclic compounds such as (di)azines,15 benzothiazole16 and imidazole.17Table 1 compares the electronic properties of selected electron withdrawing and donating groups used as A/D parts of push–pull molecules through the Hammett σp and Pytela σi substitution constants.18
| Acceptors | Donors | ||||
|---|---|---|---|---|---|
| Subst. | σ p | σ i | Subst. | σ p | σ i |
| NO2 | 0.78 | 0.606 | NMe2 | −0.83 | 0.089 |
| CN | 0.66 | 0.525 | NHMe | −0.70 | 0.094 |
| CHO | 0.42 | 0.385 | NH2 | −0.66 | 0.089 |
| COMe | 0.50 | 0.286 | NHPh | −0.56 | — |
| COPh | 0.43 | — | NPh2 | −0.22 | — |
| COOH | 0.45 | 0.264 | OH | −0.37 | 0.157 |
| COOMe | 0.45 | 0.274 | OMe | −0.27 | 0.220 |
| CF3 | 0.54 | 0.372 | OPh | −0.03 | 0.278 |
| COCF3 | 0.80 | — | SMe | 0.00 | 0.217 |
| C6F5 | 0.27 | — | 2-Thienyl | 0.05 | — |
| SO2Me | 0.72 | 0.551 | Fc | −0.18 | — |
| SO2CN | 1.26 | — | |||
| SO2CF3 | 0.96 | — | |||
| SF5 | 0.68 | — | |||
| Pyridin-4-yl | 0.44 | — | |||
| Benzoxazol-2-yl | 0.33 | — | |||
| Benzothiazol-2-yl | 0.29 | — | |||
The data in Table 1 rationalizes why substituents such as nitro/cyano and N,N-dimethylamino are among the strongest and most widely used electron acceptors and donors. Beside these groups, less common (perfluoro)sulfo(a)nyl and ferrocenyl groups also impart notable electronic effects (resonance and inductive) to push–pull molecules, as indicated by their high σp constants. Compared to N,N-dialkylamino (σp = −0.83), the increasingly popular N,N-diphenyl(aryl)amino group (σp = −0.56) possesses a lower donating ability, most likely due to a partial delocalization of the amine lone electron pair to the appended phenyls(aryls), which are generally arranged in a nonplanar manner. In modern push–pull chromophores, the π-conjugated path is created by a combination of multiple bonds (olefinic and acetylenic scaffolding)10 and aromatic (mostly benzenoids)10 or heteroaromatic spacers.15,17 Push–pull molecules may adopt several arrangements. The most widely utilized are linear (D–π–A), quadrupolar (D–π–A–π–D or A–π–D–π–A) and octupolar/tripodal ((D–π)3–A or (A–π)3–D) systems. However, push–pull molecules were also reported with extraordinary arrangements such as V- (ref. 19), Y- (ref. 17), H- (ref. 20) and X-shapes.21–23
Besides the aforementioned general use of organic π-conjugated molecules, push–pull molecules found predominant applications as chromophores with nonlinear optical (NLO) properties,24 electro-optic25 and piezochromic26 materials, NLO switches,27 photochromic28 and solvatochromic probes29 as well as active layers in DSSCs.8 The second- and third-order optical nonlinearities of push–pull molecules are mainly affected by the extent of the ICT, and thus result in pre-polarization of the π-system.30 In principle, the ICT and the corresponding HOMO–LUMO gap, optical properties and NLO response can be tailored by (i) attaching electron donors and acceptors of various electronic nature, (ii) assuring efficient D–A interaction, (iii) extension, composition and spatial arrangement of the π-linker, (iv) reducing the bond-length alternation (BLA) of the π-system and (v) planarization of the entire molecule.31 Hence, in the following text will be reviewed recent examples of push–pull chromophores. The main purpose of this article is not to comprehensively review all available push–pull molecules but rather to demonstrate the fundamental principles of property tuning on suitable examples. Structural aspects and relationships mainly affecting the energies of the frontier molecular orbitals as well as the optical properties will be discussed. The influence of such tuning on the prospective application of the given chromophore will also be considered. The property tuning will be discussed according to the part of the D–π–A system involved (donor, π-system and acceptor).
2. Tuning through the donor
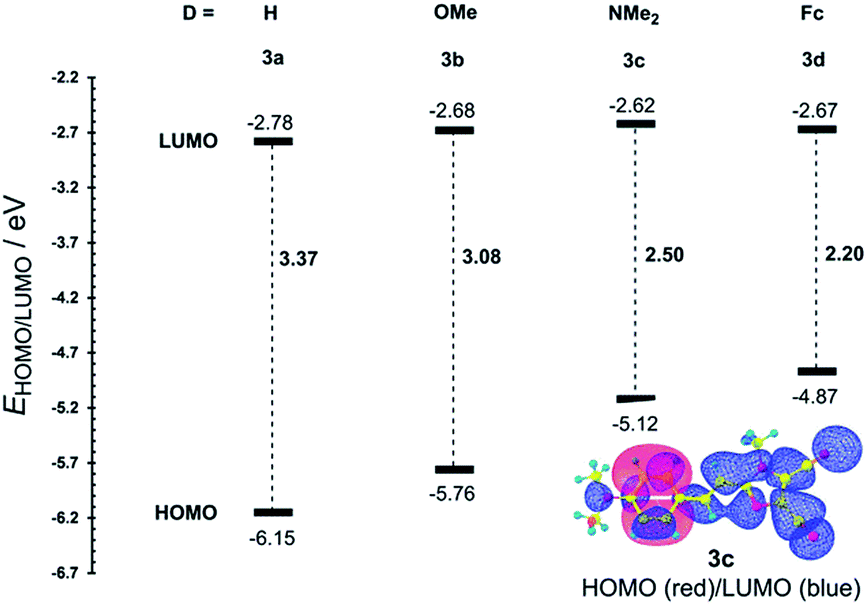
The most widely employed electron donors in push–pull molecules involve functional groups possessing +M effect such as N,N-dialkylamino (typically N,N-dimethylamino), alkoxy groups (typically methoxy) as well as auxiliary electron donors, such as electron rich five-membered heteroaromates (typically thiophene) or organometallic compounds (typically ferrocene). The molecular properties of D–π–A systems can be tailored by its replacement (stronger/weaker), addition (one or more donors) or position (conjugating/nonconjugating arrangement) of the electron donor. In 2010, we published a series of donor-substituted D–π–A molecules 1–6 bearing a 1-methylimidazole-4,5-dicarbonitrile acceptor (dicyanoimidazole (DCI), Fig. 2).32,17,30f In this systematic study, property tuning has been achieved by increasing the donating ability of the appended donors (H < MeO < Me2N) as well as the π-system extension. More recently, the series a–c have been completed by analogous compounds d bearing a ferrocenyl donor.14a The synthesis of compounds 1–6 involves Suzuki–Miyaura cross-coupling of donor substituted boronic acids or their pinacol esters33 with a 2-bromo-DCI precursor. For a demonstration of the impact of the electron donor, we can select compounds 3a–d bearing a styryl π-linker. Among others, the molecular structure and spatial arrangement of the methoxy and ferrocenyl substituted chromophores 3b and 3d are shown in Fig. 3. As can be seen, the DCI acceptor and the styryl linker adopt a planar arrangement, which assures an efficient D–A interaction. Electrochemically measured HOMO and LUMO levels and their differences Eg as well as the positions of the longest wavelength absorption maxima λmax are summarized in Table 2. However, for better understanding, the data is also presented as an energy level diagram (Fig. 4) and a UV/Vis absorption spectrum (Fig. 5).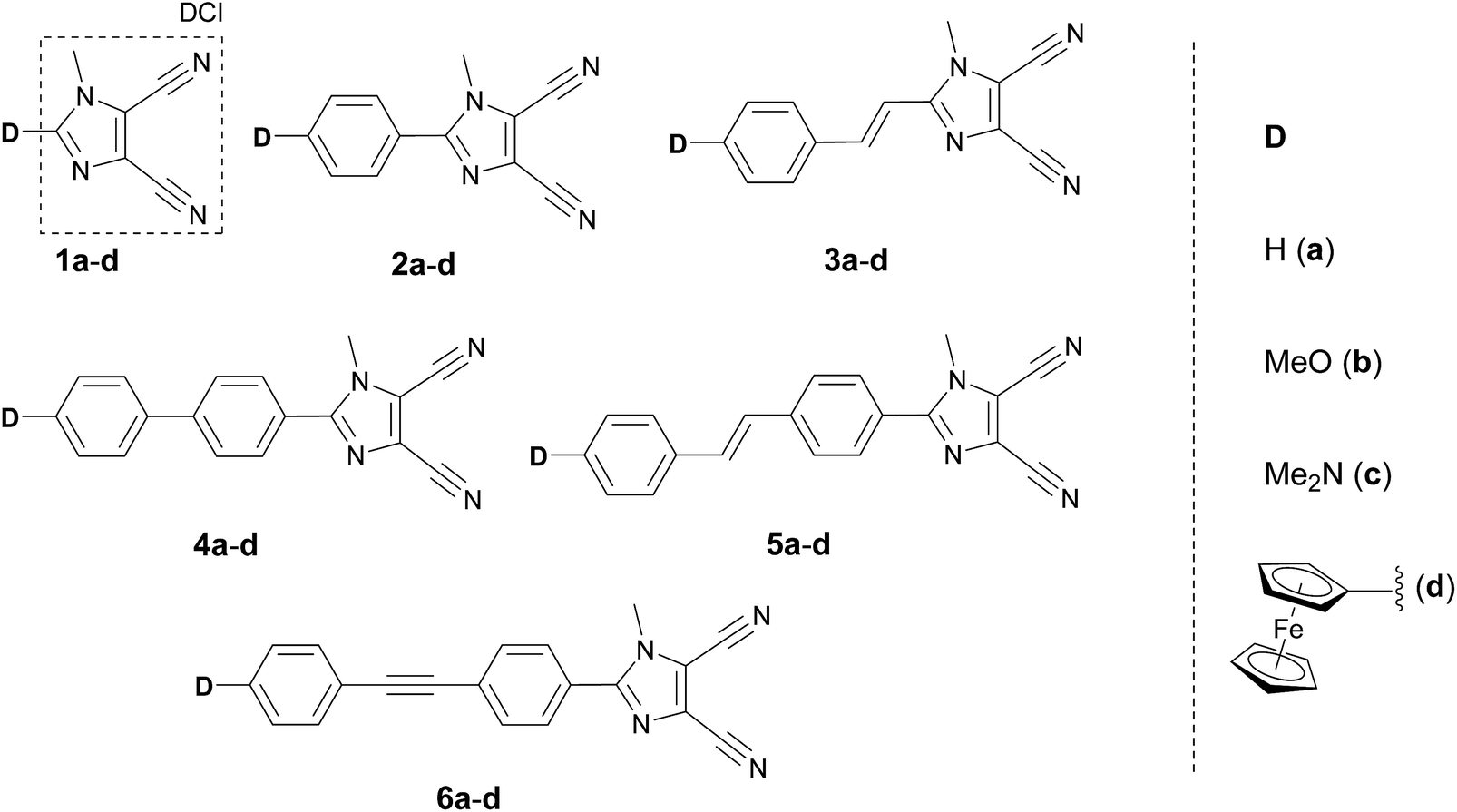 | ||
| Fig. 2 Structure of DCI-derived push–pull molecules 1–6 with various electron donors. | ||
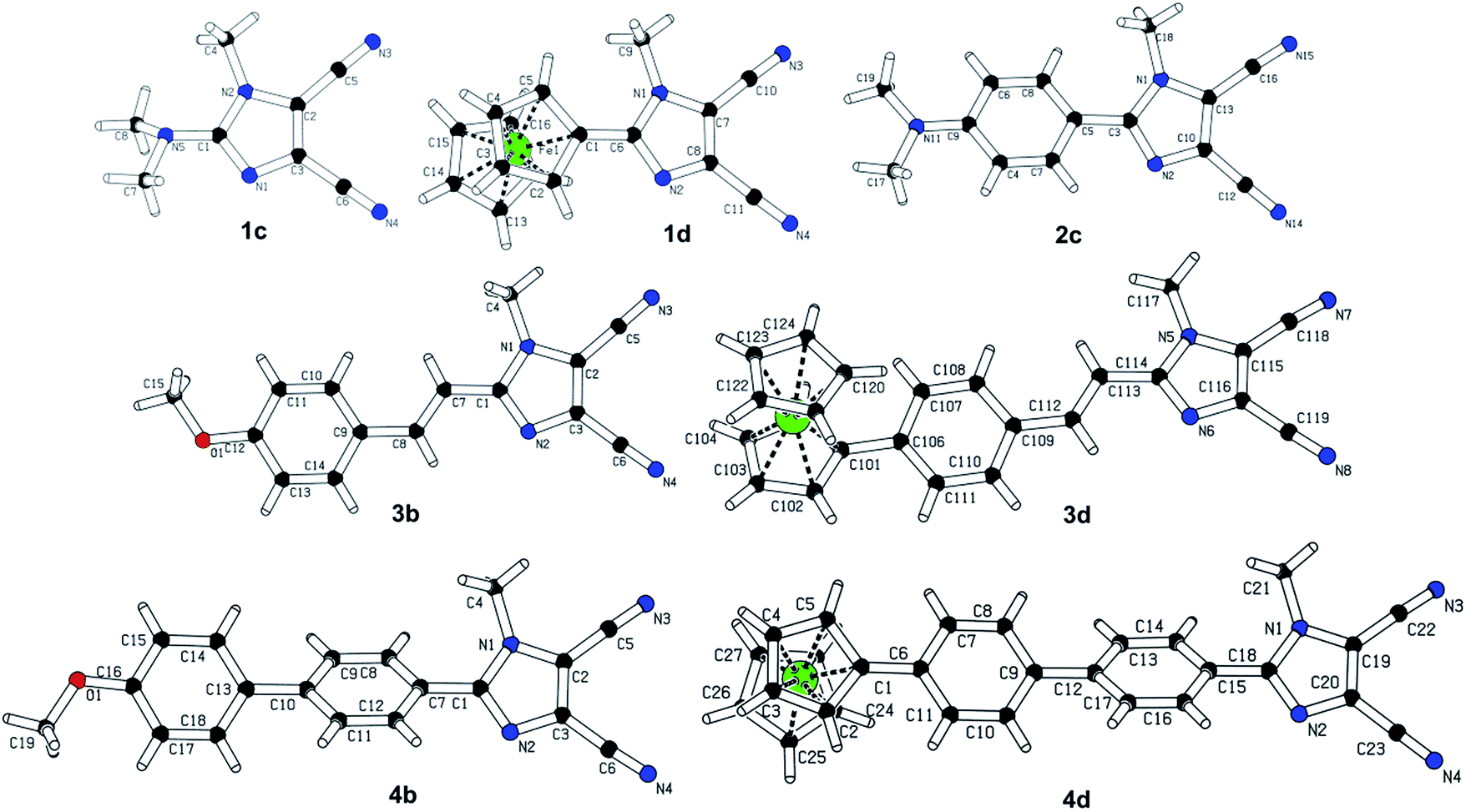 | ||
| Fig. 3 Molecular X-ray structures of successfully crystallized chromophores from the series 1–6. | ||
| Comp. | D | E HOMO [eV] | E LUMO [eV] | E g [eV] | λ max c [nm (eV)] |
|---|---|---|---|---|---|
| a Measured by CV, RDV and DC polarography in acetonitrile and recalculated.34 b Measured by CV and RDV in DMF and recalculated.34 c Measured in dichloromethane at c = 10−5 M. | |||||
| 3a | H | −6.15a | −2.78a | 3.37 | 313(3.96) |
| 3b | MeO | −5.76a | −2.68a | 3.08 | 331(3.75) |
| 3c | Me2N | −5.12a | −2.62a | 2.50 | 381(3.25) |
| 3d | Fc | −4.87b | −2.67b | 2.20 | 346(3.58)/472(2.63) |
 | ||
| Fig. 4 Energy level diagram showing HOMO/LUMO levels of 3a–d. | ||
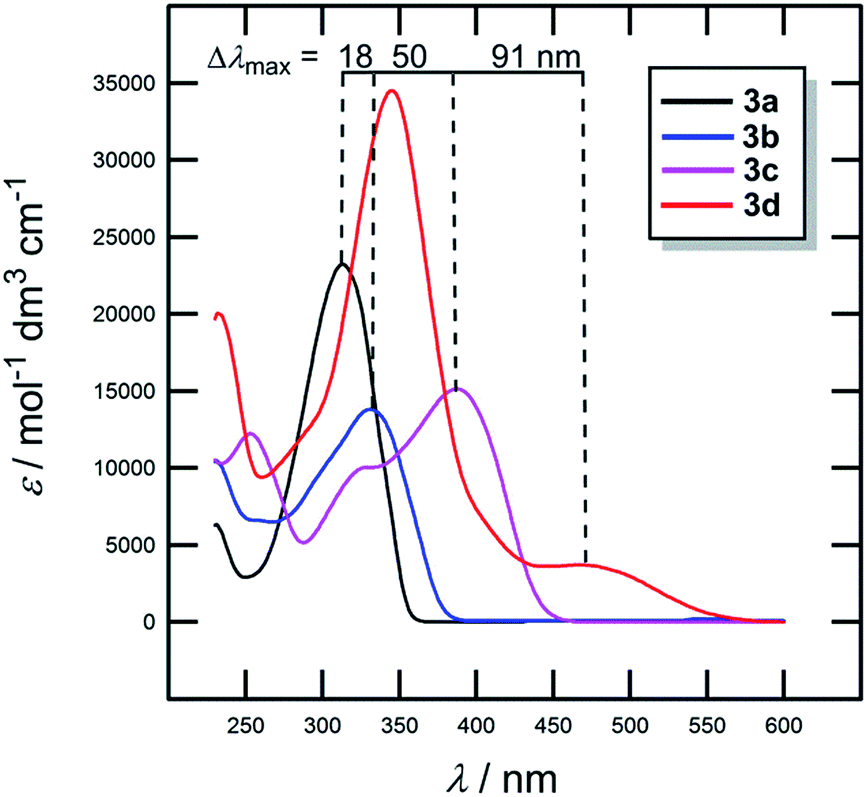 | ||
| Fig. 5 UV/Vis absorption spectra of chromophores 3a–d. | ||
While the LUMO levels remained almost unaltered (ELUMO = −2.78 to −2.62 eV), the principle changes were observed in the HOMO energies (EHOMO = −6.15 to −4.87 eV). Considering the identical acceptor and the styryl π-system in all chromophores, the observed changes clearly reflect various electronic behaviours of the appended donors. Replacement of the hydrogen in 3a by a methoxy group (3b) shifted the HOMO–LUMO gap from 3.37 to 3.08 eV (Fig. 4). Further addition of donors such as the N,N-dimethylamino (3c) or ferrocenyl (3d) groups resulted in the decrease of Eg to 2.50 and 2.20 eV, respectively. Hence, the HOMO level and thus resulting Eg of a given acceptor substituted π-system can be tuned very easily and efficiently by a variation of the donor (ΔEg = 1.17 eV). The DFT-derived HOMO and LUMO in chromophore 3c are visualized in Fig. 4.35 As expected, the HOMO is localized on the donating N,N-dimethylanilino moiety, while the LUMO is centred on the DCI acceptor and the adjacent part of the π-linker. The UV/Vis absorption spectra of 3a–d are dominated by intensive CT-bands appearing between 313 and 381(472) nm (Table 2, Fig. 5). By increasing the donating strength of the appended donors in the order H < MeO < Me2N, the position of the CT-band shifted bathochromically by 19 and 50 nm. The ferrocene donor showed two particular CT-bands. The intense high-energy band appearing at 346 nm corresponds to the HOMO-3 → LUMO interaction (cyclopentadienyl → DCI); the weak low-energy band at 472 nm originates from the interaction between the Fe-centred HOMO and the DCI-centred LUMO.14a Hence, ferrocene must be considered as a donor featuring dual ICT of various efficiencies. However, the optical properties of a push–pull chromophore can be tuned through the donor in a similar manner to their HOMO–LUMO gaps.
In the 90s, very nice examples of property tuning in push–pull molecules by variation of the donors were also demonstrated by Würthner et al.,36 Perry et al./Brédas et al.37 and Marder et al.38 Würthner demonstrated substituent variation on a 2,2′-bithiophene scaffold 7a–e end-capped with a nitro acceptor, Perry and Brédas focused experimentally/theoretically on the donor–acceptor diphenylacetylenes 8a–e and Marder showed highly polarizable push–pull imines based on 2,6-di-tert-butylindoanilines 9a–f (Fig. 6). All these push–pull molecules were investigated as efficient NLO phores (Table 3). Electron donors are represented by the hydrogen, bromo, methoxy, methylthio, amino, N-methylamino, N,N-dimethylamino and 1-pyrrolidino (PyN) groups. Optoelectronic properties of push–pull molecules 7–9 were evaluated by means of their ground state dipole moment μ, its change upon excitation Δμ, the position of the longest-wavelength absorption maxima (including solvatochromism), as well as the first molecular hyperpolarizability β0 (EFISH). As a general trend, bathochromic shift of the CT-band, raised dipole moment and its change upon excitation and increased magnitude of β0 have been observed with increasing donor strength. The most bathochromically shifted λmax and highest nonlinear optical responses were measured for push–pull molecules bearing a N,N-dimethylamino donor. In any case the high donating character of pyrolidin-1-yl as in 7e should not be neglected. Two methoxy groups attached at the conjugated positions 2 and 4 (9evs.9d) caused an additional bathochromic shift of 29 nm, however the impact on the first hyperpolarizability is diminished. The MeS donor seems to be slightly more efficient than MeO. It red-shifts the CT-band similarly to MeO, however, due to the presence of the polarizable sulphur atom, NLO phores 7c, 8b and 9c undergo large changes in the dipole moment upon excitation and are slightly more polarized than 7b, 8a and 9d (Table 3). Besides the aforementioned contributions, Marder, Brédas, Perry, Cheng and others have developed the concept of a well-balanced donor/acceptor/π-system, which proved to be very crucial for the optimization of the NLO performance of organic D–π–A systems.39
 | ||
| Fig. 6 Push–pull 2,2′-bithiophenes 7, diphenylacetylenes 8 and 2,6-ditertbutylindoanilines 9. | ||
| Comp. | D | μ g [D] | Δμ [D] | λ max [nm (eV)] | β 0 c [10−30 esu] |
|---|---|---|---|---|---|
| a Measured in benzene. b Measured in n-hexane. c EFISH experiment (1910 nm). d Measured in chloroform. e [10−30 esu]. | |||||
| 7a | H | 5.04a | 7.55 | 375(3.31)b | 9.4 |
| 7b | MeO | 6.03a | 11.35 | 408(3.04)b | 18.9 |
| 7c | MeS | 4.98a | 16.31 | 391(3.17)b | 26.9 |
| 7d | Me2N | 7.93a | 13.96 | 466(2.66)b | 41.0 |
| 7e | PyN | 8.41a | 16.47 | 499(2.48)b | 56.0 |
| 8a | MeO | 4.40 | 6.20 | 347(3.57)d | 14.0 |
| 8b | MeS | 4.00 | 5.40 | 358(3.46)d | 20.0 |
| 8c | H2N | 5.50 | 9.50 | 379(3.27)d | 24.0 |
| 8d | MeHN | 5.70 | 7.70 | 400(3.10)d | 46.0 |
| 8e | Me2N | 6.10 | 5.70 | 416(2.98)d | 46.0 |
| 9a | H | 1.5e | — | 428(2.90)d | 4.5 |
| 9b | Br | 1.3e | — | 432(2.87)d | 9.9 |
| 9c | MeS | 2.3e | — | 476(2.61)d | 11.0 |
| 9d | MeO | 2.4e | — | 469(2.64)d | 12.0 |
| 9e | 2,4-DiMeO | 3.4e | — | 498(2.45)d | 13.0 |
| 9f | Me2N | 3.9e | — | 558(2.22)d | 47.0 |
More recently, Blanchard-Desce et al. reported NLO-active donor–acceptor polyenes 10a–c and push–pull arylbithiophenes 11a–c with efficient fluorescence and two-photon absorption (Fig. 7).40 Both π-conjugated scaffolds were end-capped with N,N′-diethylthiobarbituric acid as an acceptor, while H, MeO, Me2N, N,N-dibutylaninilo, julolidine, and 5-(piperidin-1-yl)thiophene-2-yl (PIT) groups were employed as electron donors.
 | ||
| Fig. 7 Donor–acceptor polyenes 10, arylbithiophne 11 and pyrrole-pyridinium chromophores 12. | ||
Table 4 demonstrates that both absorption and emission maxima of polyenes 10 and arylbithiophenes 11 are red-shifted with increasing donor strength. However, the first hyperpolarizability of 10a–c decreased from 150 to 55 × 10−30 esu.
| Comp. | D | λ Amax a [nm (eV)] | λ Emax a [nm (eV)] | E g b [eV] | NLO |
|---|---|---|---|---|---|
| a Measured in chloroform. b Measured by CV in acetonitrile (vs. SCE). c β 0 values measured by EFISH experiment (1907 nm) in chloroform [10−30 esu]. d TPA cross-section at 1100 nm in chloroform [GM]. | |||||
| 10a | Bu2NC6H4 | 587 | — | 1.68 | 150c |
| 10b | Julolidine | 614 | — | 1.54 | 130c |
| 10c | PIT | 619 | — | 1.54 | 55c |
| 11a | H | 527 | 596 | — | — |
| 11b | MeO | 541 | 644 | — | — |
| 11c | Me2N | 588 | 781 | — | 1180d |
According to Blanchard-Desce,40a this is a consequence of reduced BLA in polyenals with the π-system length as in 10a–c (less balanced D–π–A NLO phore, for the BLA optimization see the further text). In contrast to polyenes 10, arylbithiophenes 11 showed a significant increase in fluorescence and large one- and two-photon brightness reaching the TPA cross-section of 1180 GM, measured for 11c. Hence, arylbithiophenes 11 hold promise as biphotonic fluorescent probes for bioimaging.
Tuning of the donor in TPA-active push–pull chromophores 12 has also been demonstrated by Abbotto et al. (Fig. 7).41 They have investigated simple pyridinium salts 12a–e linked to electron rich pyrrole (donor), which was further substituted by methyl and phenyl groups. The pyrrole electron releasing ability and the corresponding ICT and optical properties were finely tuned by minor structural changes, which, however, have a significant impact on frequency up-converted lasing. Hence, chromophores 12a–e were able to convert easily available infrared laser radiation at 790 nm into more useful visible laser emission over a wide region of the visible spectrum (522–576 nm) via a two-photon absorption-induced fluorescence phenomenon.
A proper selection of the donor part of the organic push–pull molecule is also currently a common strategy to improve the performance of the dye-sensitizer in DSSCs. In this respect, arylamines belong undoubtedly to the most widely employed electron-donor moieties.8 Zhang et al.42 and Jungsuttiwong et al.43 have theoretically investigated organic dyes 13 and 14 with various peripheral donors based on a combination of carbazole, triphenylamine and bisfluorenyl moieties (Fig. 8 and Table 5).
 | ||
| Fig. 8 Donor tuning in dye-sensitizers 13 and 14. | ||
| Comp. | E HOMO [eV] | E LUMO [eV] | E g [eV] | λ max [nm (eV)] |
|---|---|---|---|---|
| a DFT calculation in THF (CAM-TD-B3LYP/MPW1K). b Experimental λmax values for 13a and 13b in THF are 502(2.47) and 523(2.37) nm (eV). c DFT calculation in vacuum (CAM-TD-B3LYP/6-31 G(d,p). | ||||
| 13a | −5.29a | −2.73a | 2.56a | 481(2.58)a,b |
| 13b | −4.81a | −2.66a | 2.15a | 523(2.37)a,b |
| 13c | −4.77a | −2.67a | 2.10a | 523(2.37)a |
| 14a | −5.27c | −2.61c | 2.66c | 430(2.88)c |
| 14b | −5.15c | −2.65c | 2.50c | 429(2.89)c |
| 14c | −5.14c | −2.69c | 2.45c | 428(2.90)c |
| 14d | −4.96c | −2.57c | 2.39c | 441(2.81)c |
| 14e | −4.78c | −2.54c | 2.24c | 453(2.74)c |
Both dye-sensitizers 13 and 14 possess a good TiO2-anchoring group (cyanoacrylilic acid) and well-tuned HOMO and LUMO. Whereas the LUMO levels are sufficiently negative with respect to the conduction band of TiO2 (∼3.2 eV), the HOMO levels are close to the redox potential of the iodide/triiodide couple for efficient dye regeneration. In contrast to the recent concept of D–A–π–A systems,8a new dye-sensitizers 13 and 14 represent D–D–π–A push–pull molecules. As a general trend, we can observe that the absorption edge of the dye shifts bathochromically and the HOMO level and the corresponding Eg decreases with increasing number and electron releasing ability of the auxiliary donors. The donating ability of carbazole is generally weaker than that of triphenylamine (e.g. molecules 13avs.13b or 14cvs.14e).
3. Tuning through the π-system
The π-conjugated bridge between the electron donor and acceptor is an integral part of the D–π–A system and, therefore, its influence on the optoelectronic properties will be discussed herein. The π-linker consists of a combination of multiple bonds, aromates and heteroaromates and the basic structure–property relationships can be demonstrated on the N,N-dimethylamino substituted DCI molecules 1c–6c shown in Fig. 2. Whereas molecule 1c has the donor connected directly to the DCI acceptor, in chromophores 2c–6c both moieties are separated by an additional π-linker such as 1,4-phenylene (2c), styryl (3c), biphenyl (4c), stilbenyl (5c) and phenylethynylphenyl (6c). The X-ray analyses in Fig. 3 show that particular π-linkers adopt various spatial arrangements. The five-membered DCI unit connected directly to 1,4-phenylene as in chromophores 2 and 4 adopts a nonplanar arrangement with a torsion angle of 29–33°. Moreover, both 1,4-phenylene moieties of the biphenyl linker (4) are highly twisted. On the other hand, additional olefinic/acetylenic units as in 3, 5 and 6 caused (partial) planarization of the entire π-system. The HOMO/LUMO and λmax values of molecules 1c–6c are summarized in Table 6 and Fig. 9 and 10.| Comp. | E HOMO a [eV] | E LUMO a [eV] | E g a [eV] | λ Amax b [nm (eV)] | λ Emax b [nm (eV)] | β HRS c [10−30 esu] |
|---|---|---|---|---|---|---|
| a Measured by CV, RDV and DC polarography in acetonitrile and recalculated.34 b Measured in dichloromethane. c HRS experiment in dichloromethane (1064 nm). | ||||||
| 1c | −5.81 | −2.47 | 3.34 | 293(4.23) | — | — |
| 2c | −5.36 | −2.51 | 2.85 | 316(3.92) | 460(2.70) | 19.2 |
| 3c | −5.12 | −2.62 | 2.50 | 381(3.25) | 474(2.62) | 28.5 |
| 4c | −5.22 | −2.58 | 2.64 | 346(3.58) | 504(2.46) | 26.3 |
| 5c | −5.01 | −2.62 | 2.39 | 380(3.26) | 533(2.33) | 28.0 |
| 6c | −5.15 | −2.65 | 2.50 | 364(3.41) | 515(2.41) | 40.4 |
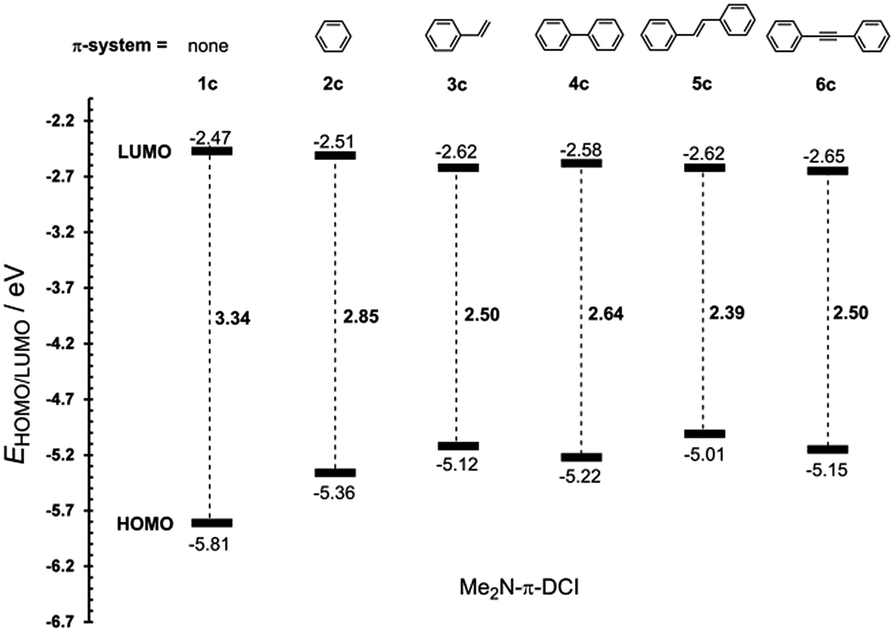 | ||
| Fig. 9 Energy level diagram of 1c–6c with systematically enlarged π-linker. | ||
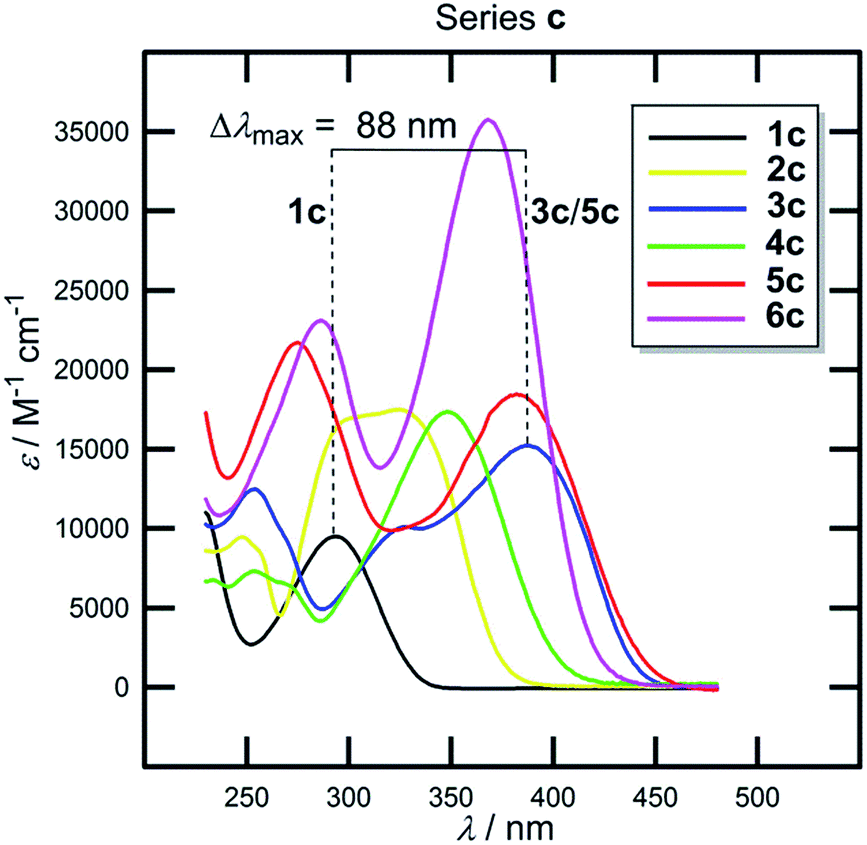 | ||
| Fig. 10 UV/Vis absorption spectra of 1c–6c. | ||
As can be seen from Fig. 9, the most significant changes are observed in the HOMO levels. Extension of the π-system by one 1,4-phenylene moiety (1c → 2c) lowered the HOMO–LUMO gap by 0.49 eV. Further insertion of an olefinic unit between the DCI and 1,4-phenylene parts of the π-linker (2c → 3c) resulted in a drop in Eg by an additional 0.35 eV. Despite being longer than styryl, the nonplanar bihenyl π-linker in 4c allows less D–A interaction and, therefore, the HOMO–LUMO gap is raised to 2.64 eV. However, the gap can be lowered by planarization of the π-linker by olefinic or acetylenic units as in 5c and 6c. A comparison of 5c and 6c also allowed a demonstration of the “transparency” of the double and triple bond. The HOMO–LUMO gap of 5c is lower by 0.11 eV compared to 6c, which leads to the assumption that, in contrast to a vacuum,44 the olefinic unit allows a higher D–A interaction than the acetylenic one. Hence, the most efficient ICT has been achieved in molecules 2c and 5c bearing fully-planar and transparent styryl and stilbenyl π-systems.
Now we can analyse the UV/Vis absorption spectra of 1c–6c (Fig. 10, Table 6). With the same donor and acceptor, all observed changes must be attributed to the π-linker.
The position of the CT-bands ranges from 293 to 381 nm and is clearly dependent on the length, composition and spatial arrangement of the π-system. Similar trends as seen by the electrochemical measurements can also be concluded from the UV/Vis spectra. Moreover, both the optical and electrochemical gaps showed a very tight correlation (Fig. S1, ESI†). Thus, in 1c–6c we can easily modulate the HOMO–LUMO gap and the position of the longest wavelength absorption maxima within the range of 0.95 eV and 88 nm by the extension and variation of the π-system between the N,N-dimethylamino donor and the DCI acceptor. Chromophores 1c–6c were used as efficient NLO phores with tunable optical nonlinearities, pH- and photo-induced NLO switches and fluorophores (Table 6).35,45
Property tuning through the extension of the π-system has also been demonstrated on polyenes and polyynes, as shown for example on molecules 10 (Fig. 7)40a and 8 (Fig. 6).37 Systematic extension of the central polye(y)ne π-system led to lower electrochemical as well as optical gaps Eg. However, it was demonstrated that the nonlinear optical response depends on the degree of BLA.39d The BLA can be defined as the difference between the average C–C single and multiple bond lengths in the conjugated backbone and arises from the mixing of limiting resonance forms (aromatic vs. quinoid, Fig. 1). Hence, the highest NLO activity was encountered for NLO phores with optimized donor–acceptor strengths and BLA (e.g.10a analogue with five double bonds possesses a β0 coefficient of 1000 × 10−30 esu, Fig. 7).40a The structure–property relationships in polye(y)nic chromophores were also intensively studied by DFT calculations and various functions and benchmarks were developed to optimize their NLO response.46
Polyenes end-capped with N,N-dibutylanilino/tetrathiafulvalene donors and dicyanovinyl/thiobarbituric acceptors 15 and 16a–c were investigated by Kelly et al.47 and Garín et al. (Fig. 11, Table 7).48 Compound 15 has been used as a model push–pull chromophore to investigate the effect of polyenic vs. aromatic π-linkers by resonance Raman intensity analysis. It was suggested that the two-state model is more appropriate for polyenic than aromatic chromophores. Chromophores 16 showed a very narrow HOMO–LUMO band gap of 1.23–1.08 eV. Extension of the polyene backbone caused notable changes in the LUMO energies (Ered,1, Table 7) and thus decreased the Eg. The observed huge hypsochromic shift with Δλmax = 70 nm is very unusual and was not explained. The very low solubility of 16a–c in nonpolar solvents and unavoidable measurements in DMSO would play a significant role. In any case the NLO response (μβ0 product) is clearly a function of the number of double bonds in the conjugated backbone.
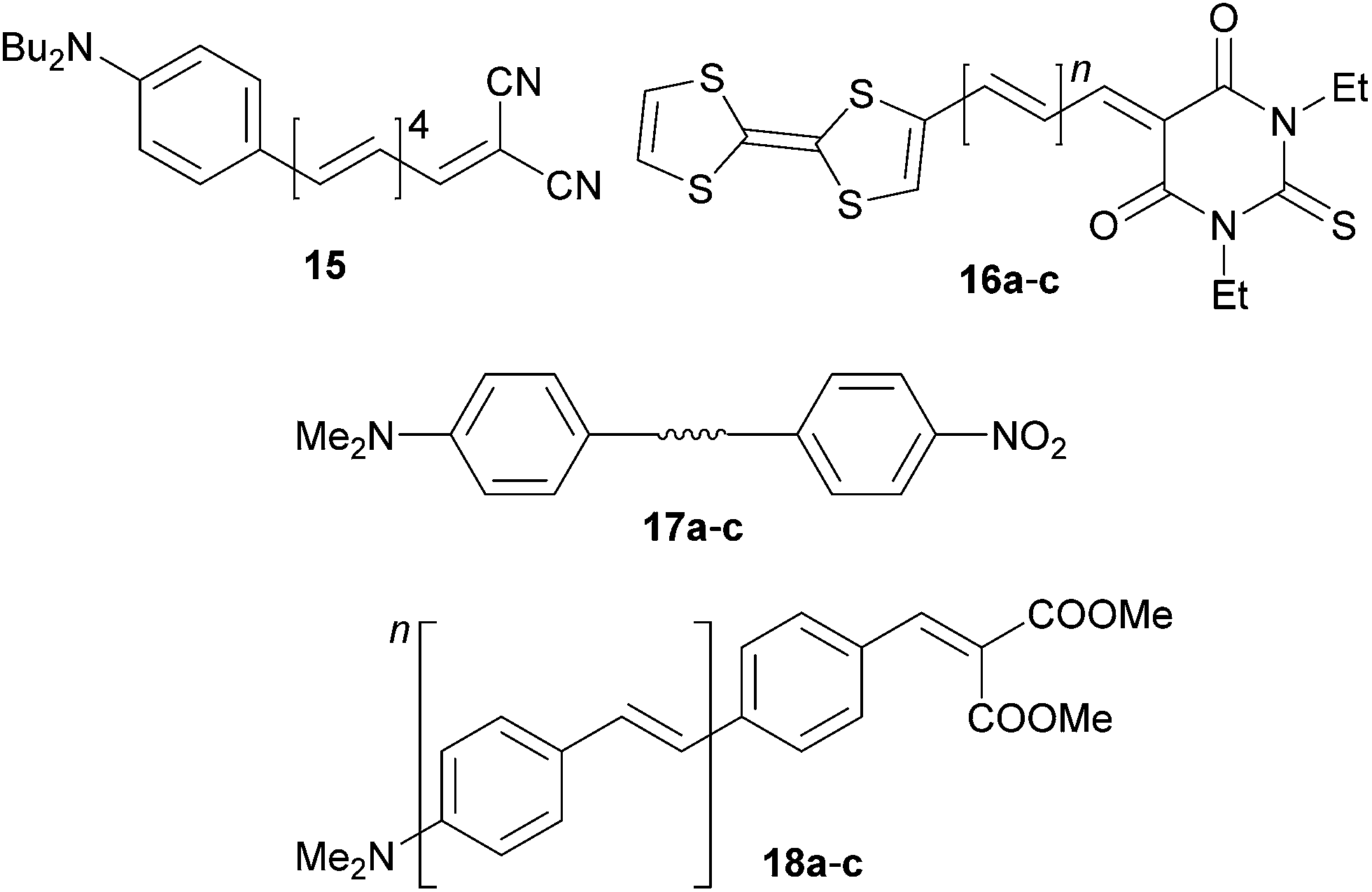 | ||
| Fig. 11 Structure of oligomeric push–pull chromophores 15–18. | ||
| Comp. | Structure | E ox,1 [V] | E red,1 [V] | E g [eV] | λ max [nm (eV)] | NLO |
|---|---|---|---|---|---|---|
| a Measured by CV vs. Ag/AgCl in CH2Cl2/DMF. b Measured in DMSO. c μβ 0 product measured by EFISH experiment (1907 nm) in DMSO [10−48 esu]. d Measured in chloroform. e β 0 measured by EFISH experiment (1907 nm) in chloroform [10−30 esu]. f Measured in toluene. g μβ 0 product measured by EFISH experiment (1907 nm) in chloroform [10−48 esu]. | ||||||
| 16a | n = 0 | 0.60a | −0.63a | 1.23 | 716(1.73)b | 180c |
| 16b | n = 1 | 0.60a | −0.57a | 1.17 | 661(1.88)b | 347c |
| 16c | n = 2 | 0.56a | −0.52a | 1.08 | 646(1.92)b | 455c |
| 17a | C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C C |
— | — | — | 416(2.98)d | 32.8e |
| 17b | CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH CH |
— | — | — | 438(2.83)d | 42.1e |
| 17c | NN |
— | — | — | 480(2.58)d | 39.9e |
| 18a | n = 0 | — | — | — | 369(3.36)f | 120g |
| 18b | n = 1 | — | — | — | 410(3.02)f | 170g |
| 18c | n = 2 | — | — | — | 418(2.97)f | 350g |
Acetylenic, olefinic and azo units are widely used features of π-linkers. Moylan et al. have reported (non)linear optical properties of simple push–pull molecules 17a–c (Fig. 11, Table 7).49 When going from 17a to 17b and 17c, the CT-bands move bathochromically but the highest NLO response (β0) has been recorded for the 17b bearing olefinic unit. The azo linker in 17c must, however, be considered as an auxochrome, which artificially red-shifts the CT-band. Despite the higher general polarizabilities of olefinic and azo π-linkers, their facile isomerization and generally lower stability also makes the acetylenic unit well-balanced in terms of polarizability, stability and synthetic accessibility (e.g. via Sonogashira cross-coupling).
Pasini et al. reported on crescent oligo-p-phenylenevinylene (PPV) type push–pull chromophores 18a–c (Fig. 11). N,N-Dimethylamino and the less common dimethylmalonate groups were used as donor and acceptor, respectively.50 As expected, the chromophores showed bathochromically shifted λmax and enhanced NLO response with increasing number of styryl units. Whereas 18a bearing the shortest π-linker showed a strong emission in the solid state, the emissions of the extended homologues 18b–c were significantly quenched by aggregation (π-stacking). High quantum yield photoluminescence can be restored in the solution (toluene).
Diederich et al. and Biaggio et al.22d,51 reported on the synthesis and third order optical nonlinearities of X-shaped donor-substituted cyanoethynylethenes 19 (Fig. 12). Property tuning has been achieved by a systematic extension of the central π-conjugated backbone by olefinic and acetylenic units and additional dicyanovinyl (DCV)30f acceptor moieties.
 | ||
| Fig. 12 Donor-substituted cyanoethynylethenes 19 (DsCEEs). | ||
As can be seen from the data given in Table 8, an extension of the π-system in DsCEEs reduces the LUMO level (Ered,1). Hence, the electrochemically measured HOMO–LUMO gap decreases from 2.55 to 1.49 eV. A beneficial role of an additional acetylene unit (n = 1, Fig. 12) separating N,N-dimethylanilino (DMA) donors from the central conjugated backbone and causing planarization of the entire π-system is also noticeable (compare for instance 19a/19b or 19c/19d). Introduction of an additional DCV substituted branch (k = 1) as in 19g and 19h resulted in a further drop in the Eg to 1.49 eV. The optical band gap is red-shifted from 2.86 eV (434 nm) to 1.77 eV (700 nm). Thus, extension of the parent CEE scaffold in 19a/19b by properly placed acetylenic and olefinic units caused huge tuning of Eelchem/opticalg by 1.06/1.09 eV (for the correlation of both gaps see Fig. S2, ESI†). Third-order polarizabilities γrot measured by degenerate four-wave mixing showed very similar trends as seen for electrochemical and optical gaps. X-shaped chromophore 19h with two DMA and DCV units showed the third-order NLO response of 45 ± 20 × 10−23 m5 V−2 kg−1, which is one of the largest intrinsic second hyperpolarizabilities ever measured.30a
| Comp. | E ox,1 a [V] | E red,1 a [V] | E g [eV] | λ max b [nm (eV)] | γ rot c [10−23 m5 V−2 kg−1] |
|---|---|---|---|---|---|
| a Measured by CV/RDV vs. Fc+/Fc couple in dichloromethane. b Measured in dichloromethane. c Third-order polarizability (second hyperpolarizability) measured by DFWM experiment (1500 nm) in dichloromethane. d Hidden peak maxima (deconvolution). | |||||
| 19a | 0.65 | −1.90 | 2.55 | 434(2.86) | 2 ± 2 |
| 19b | 0.60 | −1.34 | 1.94 | 524(2.37) | 14 ± 3 |
| 19c | 0.52 | −1.71 | 2.23 | 498(2.49) | 4 ± 3 |
| 19d | 0.53 | −1.25 | 1.78 | 534(2.32) | 18 ± 5 |
| 19e | 0.40 | −1.18 | 1.58 | 580(2.14)d | 13 ± 3 |
| 19f | 0.46 | −1.07 | 1.53 | 600(2.07)d | 33 ± 15 |
| 19g | 0.51 | −1.38 | 1.89 | 632(1.96) | 8 ± 4 |
| 19h | 0.53 | −0.96 | 1.49 | 700(1.77)d | 45 ± 20 |
Brédas et al.52 have theoretically investigated the influence of the nature of the π-linker on two-photon absorption of quadrupolar molecules 20a–f (Fig. 13, Table 9). The cyano/N,N-dimethylamino acceptors/donors were appended to the π-conjugated backbones comprising of phenylenevinylene (20a and 20d–e), thienylenevinylene (20c), polyene (20b) and indenofluorene (20f) units. Table 9 summarizes one- and two-photon absorption properties (transition dipoles and energies). The calculated TPA cross-sections were dominantly dependent on the ground state polarization given by the quantities M and E. In general, molecules with large transition dipoles and lower transition energies delivered a stronger TPA response. Starting from 20a (stilbene π-linker), switching to the polyenic π-system (20b), less aromatic thiophenes (20c) or elongation/planarization of the π-linker (20e) allows for an increase in δ by a factor of 2–3. On the other hand, extension or geometry restrictions of the π-system by two additional acetylenic units (20d) or indenofluorene backbone (20f) have a diminished effect.
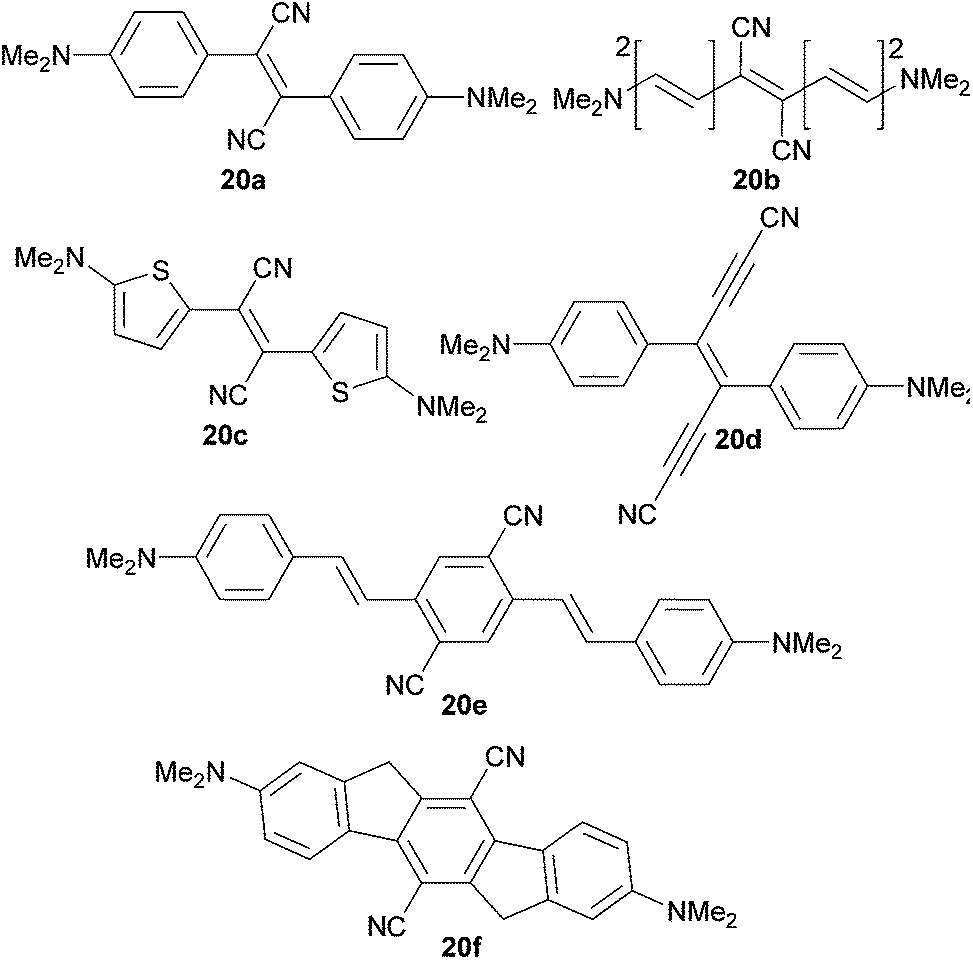 | ||
| Fig. 13 Two-photon absorbing quadrupolar molecules 20a–f. | ||
| Comp. | M ge a [D] | E ge a [eV] | M ee′ b [D] | E ge′ c [eV] | δ approx d [GM] |
|---|---|---|---|---|---|
| a Transition dipoles/energies between the ground state (g) and intermediate one-photon state (e). b Transition dipoles between one- (e) and two-photon (e′) states. c Transition energies between ground state (g) and two-photon (e′) states. d TPA cross-section. | |||||
| 20a | 8.51 | 3.22 | 12.85 | 3.79 | 512 |
| 20b | 10.52 | 3.08 | 13.96 | 4.00 | 1560 |
| 20c | 9.28 | 2.65 | 11.91 | 3.59 | 1119 |
| 20d | 8.06 | 3.14 | 11.07 | 3.75 | 365 |
| 20e | 11.27 | 3.34 | 17.22 | 3.87 | 1486 |
| 20f | 7.53 | 3.41 | 12.40 | 4.12 | 429 |
4. Heteroaromatic π-systems
Besides the commonly used aromates (benzenoids) and multiple bonds, the π-conjugated backbone of push–pull molecules can also be constructed from heteroaromates. In recent years, D–π–A systems based on heteroaromatic scaffolding have attracted considerable attention of organic and material chemists.15,17,53 It has been realized that incorporation of a heterocyclic moiety into the chromophore backbone leads to the following:• higher chemical and thermal robustness
• auxiliary electron donor or acceptor effects
• considerably enhanced polarizability
• conformational stability
• non-centrosymmetry
• acid–base and chelating properties
• easy synthesis and further modification
• improved solubility
• possible biological properties
In this respect, thiophene is undoubtedly among the most explored five-membered heterocyclic moieties, and has already found manifold applications in modern organic electronics and photonics.11 In 2012, we have investigated the extent of D–A interaction over the two most popular π-conjugated units – 1,4-phenylene and 2,5-thienylene π-linkers.54 A series of fifteen model compounds bearing a ferrocene donor and nitro acceptor have been synthesized and further investigated by crystallography, electrochemistry, UV/Vis spectra and calculations. In the chosen chromophores 21a–c (Fig. 14), the 1,4-phenylene units were systematically replaced with the 2,5-thienylene ones.
 | ||
| Fig. 14 Fc-π-NO2 model push–pull molecules. | ||
In contrast to the raised HOMO level caused by the extension and planarization of the π-system (see Fig. 9), gradual replacement of 1,4-phenylene moieties with 2,5-thienylene ones mainly lowered the LUMO levels (Fig. 15, Table 10). Hence, replacement of the first 1,4-phenylene unit resulted in a decrease of Eg from 1.51 to 1.34 eV. Replacement of the second 1,4-phenylene unit affected the HOMO–LUMO gap only negligibly. However, going from 21a → 21b → 21c, the absorption spectra (Fig. 16) showed a gradual red-shift by 52/28 and 24/29 nm. Moreover, the intensity (ε) of the weakly observed LE band (Fe → NO2/HOMO → LUMO transition) in 21a increased by 3200 and 5700 M−1 cm−1, respectively. Chromophore 21c, bearing two thiophene rings with the highest ground-state and vertically-excited state dipole moments showed the most bathochromically shifted and pronounced CT-band with λLEmax = 525 nm.
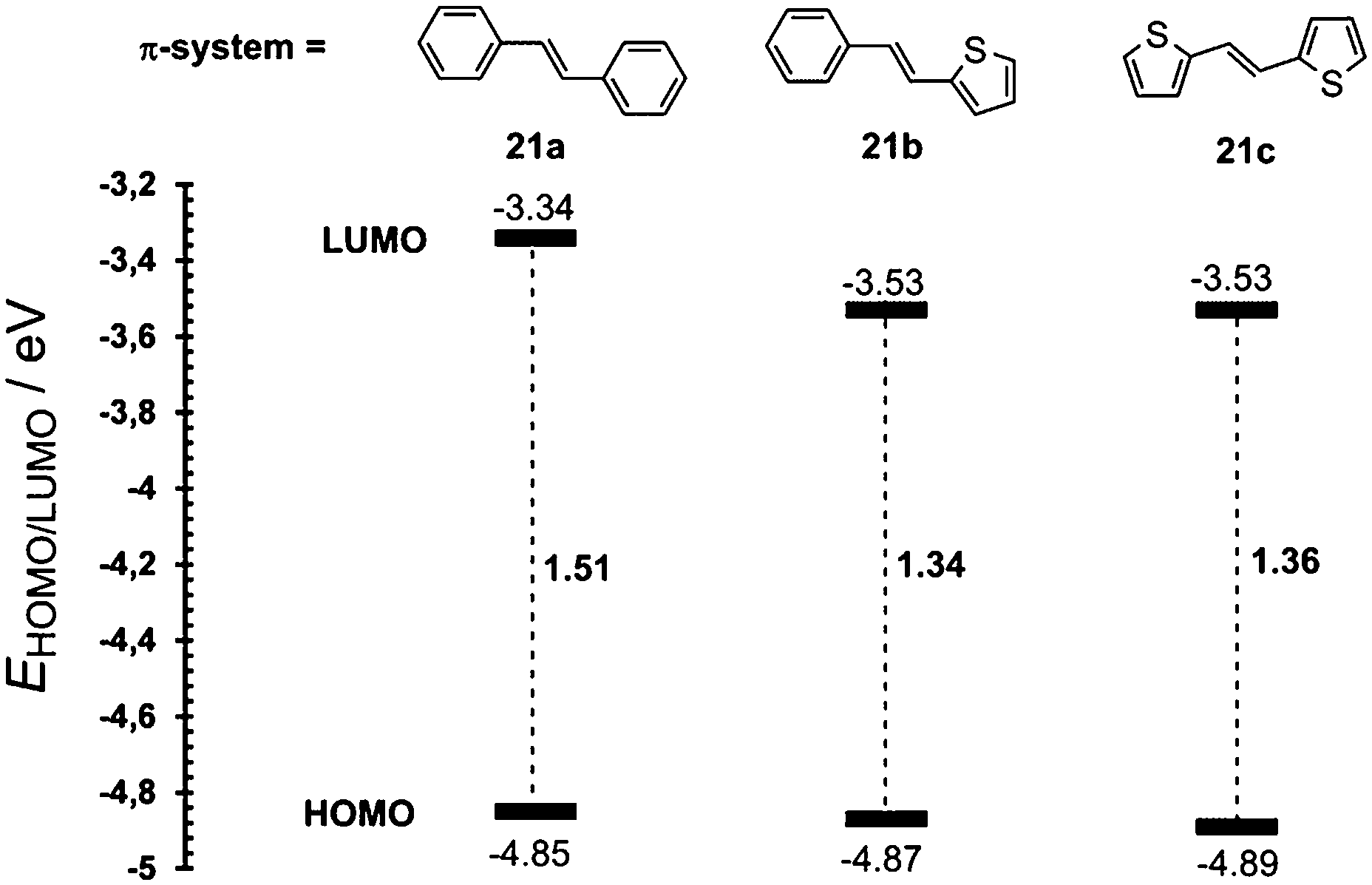 | ||
| Fig. 15 Energy level diagram of 21a–c. | ||
| Comp. | E HOMO a [eV] | E LUMO a [eV] | E g a [eV] | λ HE/LEmax b [nm (eV)] | μ gs c [D] | μ ves c [D] |
|---|---|---|---|---|---|---|
| a Measured by CV, RDV and DC polarography in acetonitrile and recalculated.34 b Position of the high- and low-energy CT-bands in dichloromethane. c DFT-derived ground-state/vertically excited-state dipole moments (M06/6-31G(df,p). | ||||||
| 21a | −4.85 | −3.34 | 1.51 | 374(3.32)/472(2.63) | 9.08 | 11.53 |
| 21b | −4.87 | −3.53 | 1.34 | 426(2.91)/496(2.50) | 10.77 | 14.91 |
| 21c | −4.89 | −3.53 | 1.36 | 454(2.73)/525(2.36) | 11.83 | 18.26 |
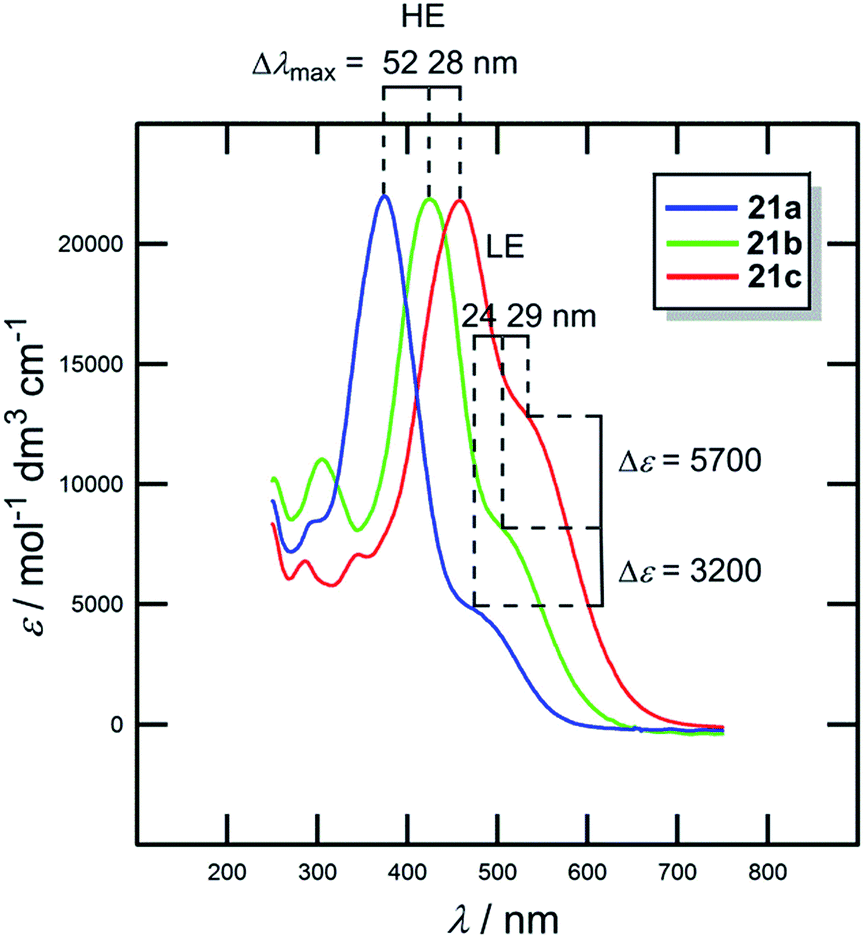 | ||
| Fig. 16 UV/Vis spectra of chromophores 21a–c in dichloromethane. | ||
Thiophene as a part of the π-system of push–pull molecules has also been investigated by Rao et al.55 Dialkylamino and DCV end-capped push–pull molecules 22a–f (Fig. 17) were investigated as tunable NLO phores. The property tuning has been achieved either by replacement of one or both 1,4-phenylene moieties in the parent molecule 22a (→ 22c → 22f) or by oligomerization of the vinylenethienylene moiety as in molecules 22b–e. Both strategies led to significant red shift as well as increased NLO response (Table 11). It is worth mentioning that gradual addition of the vinylenethienylene moiety and observed increase in the NLO response in 22b–e showed very tight linear correlation with the number of π-electrons (nπ) localized between the N,N-diethylamino donor and the DCV acceptor (Fig. S3, ESI†). In contrast to benzenoids, no NLO saturation was observed in the NLO phores bearing vinylenethienylene π-linkers. A similar set of push–pull molecules has also been experimentally and theoretically investigated by Wong et al.56 with the same outcome.
 | ||
| Fig. 17 Thiophene as part of the π-linker. | ||
| Comp. | μ gs [D] | Δμ [D] | λ max [nm (eV)] | NLO |
|---|---|---|---|---|
| a Measured in dioxane. b μβ 0 product measured by EFISH experiment (1907 nm) in dioxane [10−48 esu]. c Measured in chloroform. d Difference between the experimentally obtained ground-state and calculated excite-state dipole moments. e β SHG measured by EFISH experiment (1907 nm) in chloroform [10−30 esu]. | ||||
| 22a | — | — | 468(2.65)a | 1100b |
| 22b | — | — | 419(2.96)a | 300b |
| 22c | — | — | 513(2.42)a | 1300b |
| 22d | — | — | 547(2.27)a | 2300b |
| 22e | — | — | 556(2.23)a | 3800b |
| 22f | — | — | 584(2.12)a | 2600b |
| 23a | 10.8c | 7.4d | 719(1.72)c | 867e |
| 23b | 12.2c | 7.0d | 708(1.75)c | 686e |
In 2004, Moylan et al.57 have reported on two isomeric chromophores 23a–b (Fig. 17). The effect of the thiophene position within a D–π–A molecule has been studied. It was found that the thiophene on the donor side as in 23b produces a larger dipole moment; however, the isomer 23a with thiophene on the acceptor side showed a higher SHG response and has a higher figure of merit for NLO device applications (Table 11).
Raposo et al.58 have investigated structure–property relationships in push–pull oligothiophenes 24a–c both experimentally and theoretically (Fig. 18). The property tuning has been achieved by gradual elongation of the central oligothiophene scaffold end-capped with the PIT donor and the DCV acceptor. In contrast to the steady bathochromic shift seen in vinylenethienylene chromophores 22b–e (Table 11), the longest-wavelength absorption maxima in 24a–c showed the maxima for terthiophene 24b (λmax = 569 nm, Fig. 18). Push–pull chromophore 24b showed the NLO response (μβ) of 1150 × 10−48 esu, which is less than half of that measured for the extended analogous 22f (2600 × 10−48 esu, Table 11).59 Hence, planarization of the oligothiophene backbone by an additional olefinic unit plays a very crucial role in tuning the (non)linear optical properties.
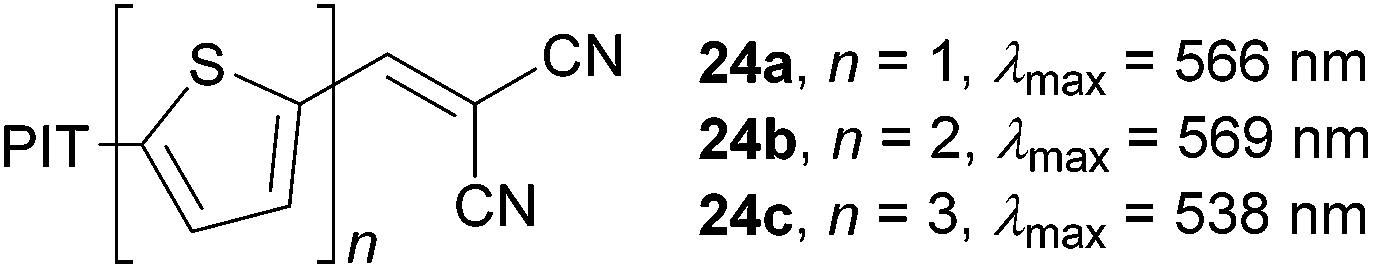 | ||
| Fig. 18 PIT-oligothiophene-DCV chromophores (λmax in chloroform). | ||
Auxiliary donor–acceptor effects of (hetero)cyclic rings incorporated into the π-linker were theoretically studied by Ratner et al.60 The NLO responses were studied in terms of electron excessivity/deficiency of the appended (hetero)ring as well as its aromaticity. Two model D–π–A patterns were investigated in which the NH2 donor and the NO2 acceptor were connected to the five-membered electron rich (hetero)cycle and the phenyl ring (25a–f) and vice versa (26a–f, Fig. 19). The calculated properties summarized in Table 12 allow us to distinguish the electron density effects and aromaticity. Whereas the most bathochromically shifted λmax (588 nm) and largest NLO response (237.8 × 10−30 esu) were calculated for 25b, with the NH2 donor appended to the electron rich cyclopentadienyl (Cp), the least electron excessive/most aromatic thiophene (25f) showed a hypsochromically shifted CT-band (402 nm) and weak NLO response (43.9 × 10−30 esu). On the other hand, the most electron excessive Cp ring in 26b showed a strongly hypsochromically shifted CT-band (440 nm) and extremely small NLO coefficient (14.7 × 10−30 esu). The largest β value (45.0 × 10−30 esu) within series 26 has been calculated for compound 26f bearing the aromatic thiophene. Hence, the arrangement of the electron donors and acceptors around the π-conjugated backbone must be well considered when designing a push–pull molecule. Similar relations can be concluded also from the theoretical work of Chandrasekhar et al.61 Namely, the NLO response of a D–π–A molecule depends strongly not only on the nature but also on the location of the heterocyclic ring within the π-linker. For example, a better NLO response can be achieved by placing the electron excessive pyrrole ring close to the D part. The position of a furane unit has a diminished effect, while the aromatic thiophene ring placed close to the A part leads to an enhanced β coefficient.
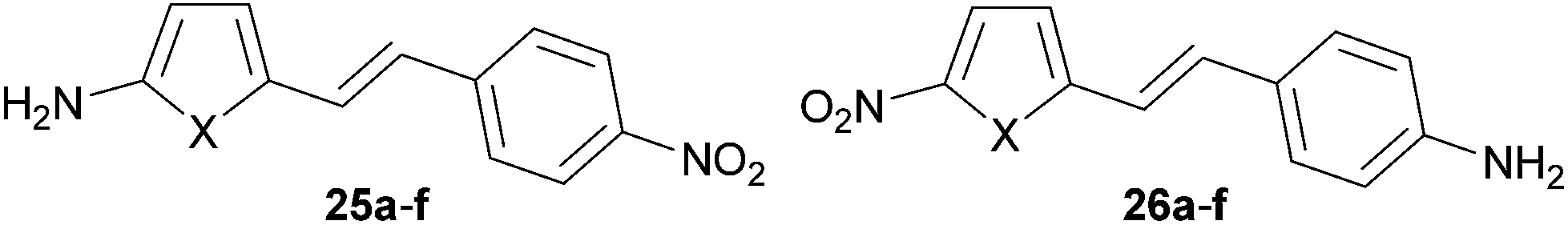 | ||
| Fig. 19 Two D–π–A patterns in molecules 25 and 26. | ||
| Comp. | X | Δμ [D] | λ max [nm (eV)] | β b [10−30 esu] |
|---|---|---|---|---|
| a The calculations were carried out using INDO/1 semiempirical Hamiltonian. b For optimized geometries. | ||||
| 25a/26a | CH2 | 14.2/11.1 | 414(3.00)/426(2.91) | 51.5/37.4 |
| 25b/26b | CH− | 12.2/3.8 | 588(2.11)/440(2.82) | 237.8/14.7 |
| 25c/26c | NH | 15.6/10.5 | 418(2.97)/417(2.97) | 55.7/33.5 |
| 25d/26d | O | 13.7/10.7 | 410(3.02)/424(2.92) | 46.6/37.5 |
| 25e/26e | PH | 15.0/8.4 | 402(3.08)/434(2.86) | 45.9/39.2 |
| 25f/26f | S | 14.0/10.7 | 402(3.08)/424(2.92) | 43.9/45.0 |
| 25d/26d | S | 14.0/10.7 | 402(3.08)/424(2.92) | 43.9/45.0 |
The length, composition and arrangement of the π-linker play a crucial role, not only for the NLO phores. Since the first Ru-sensitized DSSC reported by Grätzel in 1991,62 push–pull molecules have also found wide applications as pure (non-Ru) organic photosensitizers (donors). More recently, it has also been realized that optical properties, HOMO/LUMO gap, chemical/thermal stability and energy conversion efficiencies of a DSSC can efficiently be affected by alternation of the π-linker.63
A series of thienylene-derived π-conjugated molecules, 27a–e, which feature a N,N-diphenylamino donor and cyanoacrylic acid acceptor, and a systematically extended π-linker, has been synthesized by Hagfeldt et al. (Fig. 20).64 A systematic extension and planarization of the π-conjugated path as in the series 27a → 27b → 27d → 27c → 27e led to gradual decrease of the HOMO/LUMO energies as well as to bathochromic shift of the longest-wavelength absorption maxima up to 463 nm (Table 13). However, the highest performance has been measured for the DSSC based on 27d (η = 3.08%) The longer π-linkers as in 27c or 27e give increased spectral response (broad IPCEs) but also show pronounced losses (recombination of the electrons to the triiodide).
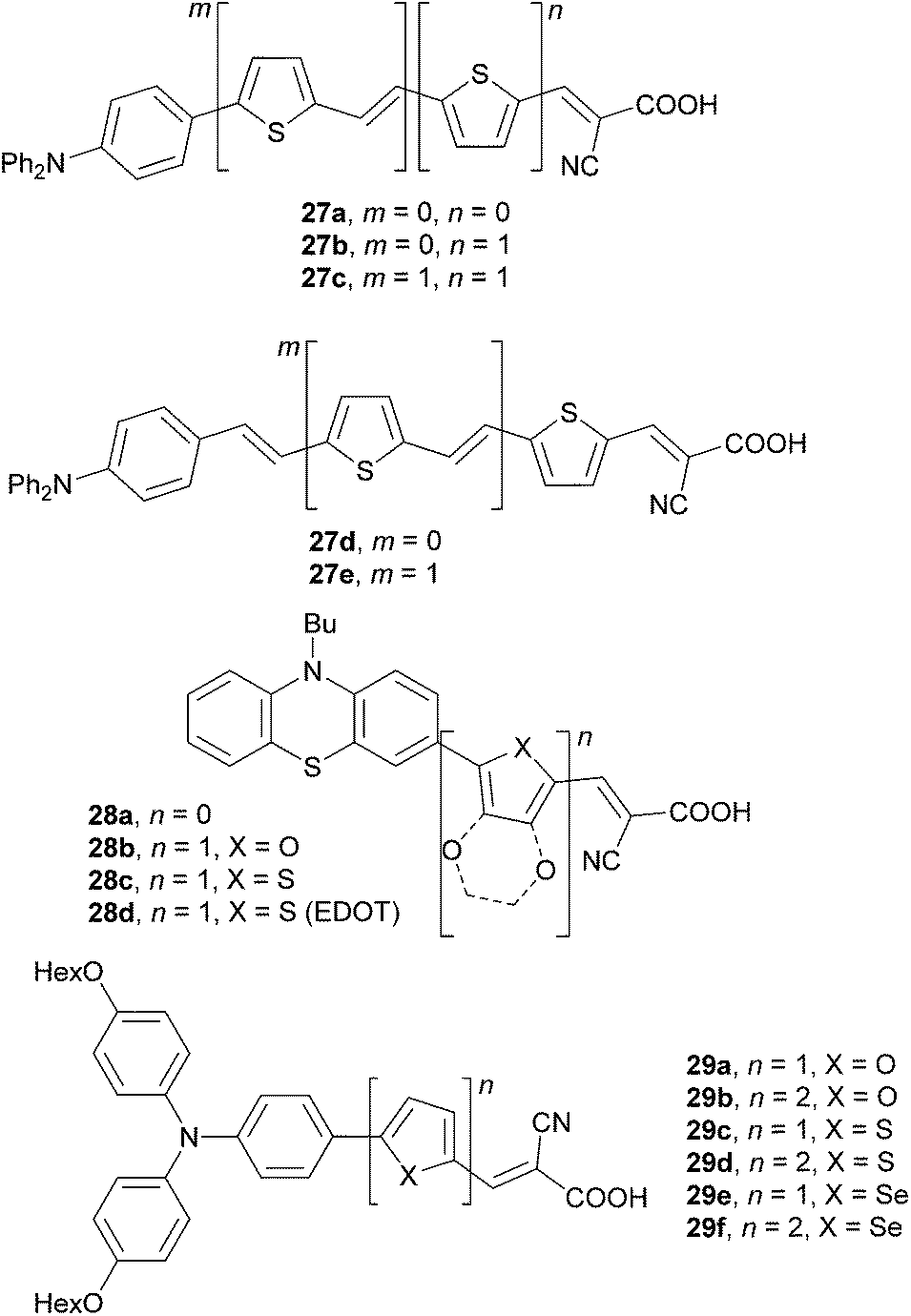 | ||
| Fig. 20 Push–pull sensitizers 27–29 with various π-linkers. | ||
| Comp. | E ox,1 [V] | E 0-0 [eV] | E LUMO [V] | λ Amax [nm (eV)] | λ Emax [nm (eV)] | η DSSC [%] |
|---|---|---|---|---|---|---|
| a Measured by DVP in acetonitrile vs. NHE (Fc/Fc+). b Transition energy estimated from the intersection of the normalized absorption and emission spectra. c E LUMO = Eox,1 + E0-0. d Absorption and emission maxima measured in acetonitrile. e Measured by CV in DMF vs. NHE (Fc/Fc+). f Estimated from the onset wavelength of the absorption spectra. g Absorption and emission maxima measured in dichloromethane. h HOMO and LUMO energies and HOMO–LUMO gaps measured by ultramicroelectrode square-wave voltammetry (vs. vacuum). i Absorption and emission maxima measured in chloroform. | ||||||
| 27a | 1.37a | 2.90b | −1.53c | 373(3.32)d | 509(2.44)d | 1.55 |
| 27b | 1.21a | 2.64b | −1.43c | 404(3.07)d | 548(2.26)d | 2.75 |
| 27c | 1.07a | 2.38b | −1.32c | 445(2.79)d | 621(2.00)d | 2.73 |
| 27d | 1.13a | 2.48b | −1.36c | 427(2.90)d | 594(2.09)d | 3.08 |
| 27e | 1.01a | 2.35b | −1.34c | 463(2.68)d | 644(1.93)d | 1.70 |
| 28a | 1.10e | 2.37f | −1.27c | 452(2.74)g | 592(2.09)g | 5.29 |
| 28b | 0.78e | 2.26f | −1.48c | 442(2.81)g | 641(1.93)g | 6.58 |
| 28c | 0.89e | 2.24f | −1.35c | 448(2.77)g | 634(1.96)g | 6.32 |
| 28d | 0.87e | 2.20f | −1.33c | 466(2.66)g | 646(1.92)g | 6.09 |
| 29a | −3.35h | 1.74h | −5.09h | 506(2.45)i | 689(1.80)i | 7.17 |
| 29b | −3.41h | 1.62h | −5.03h | 522(2.38)i | 716(1.73)i | 7.27 |
| 29c | −3.42h | 1.67h | −5.09h | 514(2.41)i | 715(1.73)i | 6.88 |
| 29d | −3.48h | 1.57h | −5.05h | 523(2.37)i | 723(1.71)i | 7.67 |
| 29e | −3.43h | 1.66h | −5.09h | 525(2.36)i | 720(1.72)i | 7.15 |
| 29f | −3.53h | 1.53h | −5.06h | 549(2.26)i | 772(1.61)i | 7.77 |
Phenothiazine dyes 28a–d were investigated by Kim et al. and Hagfeldt et al. (Fig. 20).65 The π-linker has been tailored by incorporation of furan, thiophene and 3,4-ethylenedioxythiophene (EDOT) units. Taking dye 28a as a standard, the phenothiazine donor linked to a cyanoacrylic acid acceptor through the aforementioned three π-linkers resulted in a small band gap and red-shifted absorption. Sensitizer 28b, featuring furan within the π-system, showed the highest solar energy-to-electricity conversion efficiency η = 6.58%, which is 24% higher than that achieved for 28a. DSSC based on chromophores 28c/d with thiophene and electron excessive EDOT showed lower efficiencies and overall performance.
Wang et al. have studied the effect of five-membered heteroaromates such as (bi)furan, (bi)thiophene and (bi)selenophene incorporated into the backbone of push–pull dyes 29a–f on the DSSC performance (Fig. 20).66 The data in Table 13 nicely demonstrates the impact of the extension and heteroatom replacement within the π-linker on the electrochemical HOMO–LUMO gap and optical properties, as well as the DSSC efficiencies. In accordance with the previous work of Hagfeldt and Kim, dyes featuring furan as part of the π-linker generally showed higher efficiencies than those with thiophene (compare for instance pairs of chromophores 29a/29c and 29b/29d, Table 13). Selenophene showed even better performance than furan (e.g.29e/fvs.29a/b). Sensitizers 29b, 29d and 29f with extended π-linkers comprising two consecutive heterocyclic units (n = 2) performed generally better in DSSCs than the analogous 29a, 29c and 29e (Table 13). In contrast to the application of push–pull molecules as NLO phores, their performance in DSSCs is less predictable. However, push–pull molecules bearing furan as a part of the π-system seem to be more promising compared to the thiophene analogues.
5. Proaromatic π-systems
Electronic polarization, arising from an applied electric field, of push–pull molecules bearing a ground-state aromatic π-linker (e.g. 4-nitroaniline, Fig. 1), is impeded by the concomitant loss of the resonance stabilization. Hence, the effective D–A coupling (ICT process) of a given donor/acceptor pair is suppressed, and formation of the quinoid/zwitterionic structure is rather disfavoured. As a consequence, such molecules generally deliver weak NLO responses. In 1991, Marder et al. came up with a new approach for optimizing the hyperpolarizability of π-conjugated molecules.67 This concept, later named as proaromatic π-linkers/spacers, has recently gained considerable attention as an important tool to achieve property tuning in D–π–A\D–Q–A systems. Marder has originally demonstrated the impact of such structural changes on N,N-dimethylaminonitrostilbene 30 (DANS, Fig. 21) vs. N,N-dimethylaminoindoaniline 31 (DIA, Fig. 21).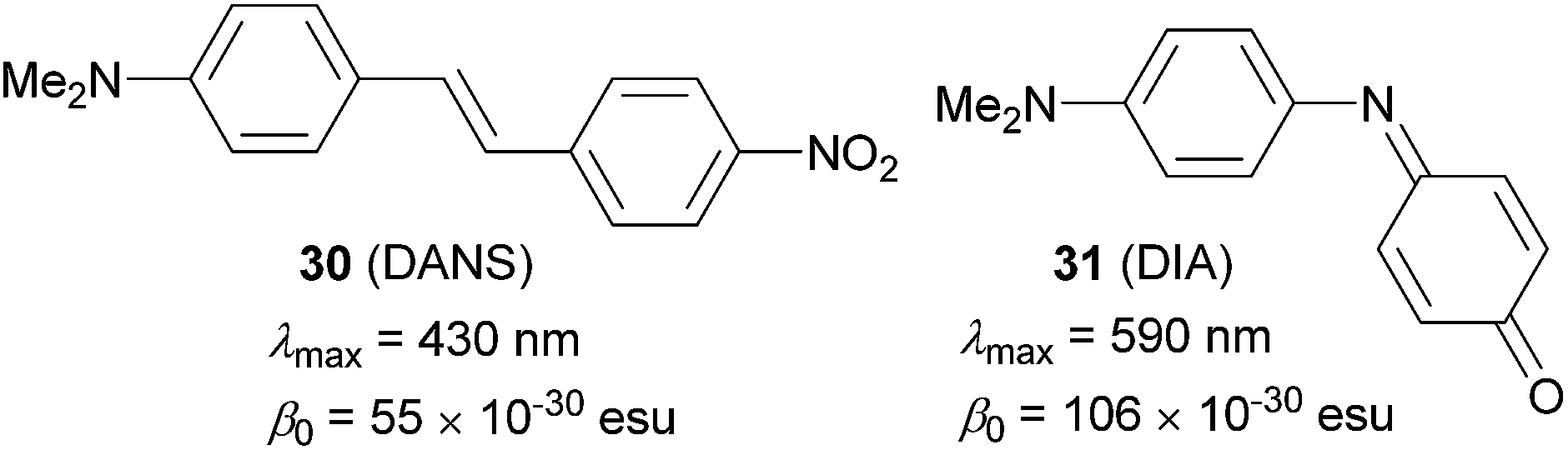 | ||
| Fig. 21 Marder’s demonstration of (pro)aromatic spacers in DANS and DIA (data measured in chloroform). | ||
Considering DANS as a standard NLO dye,68 the absorption maxima and β0 coefficient of DIA were significantly red-shifted and almost two times higher, respectively. With the same DMA donors, this effect must be attributed to the proaromatic (quinoid) indoaniline acceptor, which gains aromaticity upon excitation (reversal to the resonance forms shown in Fig. 1). In fact, either the acceptor/donor or both quinoid parts may be present in the currently known proaromatic push–pull molecules.
Diederich69et al. have shown tripodal push–pull molecules 32a–d bearing a proaromatic withdrawing moiety structurally similar to 31. However, due to the presence of an additional DCV unit, it represents an even stronger electron acceptor (Fig. 22).69 Molecule 32a has also been reported by Oda.70
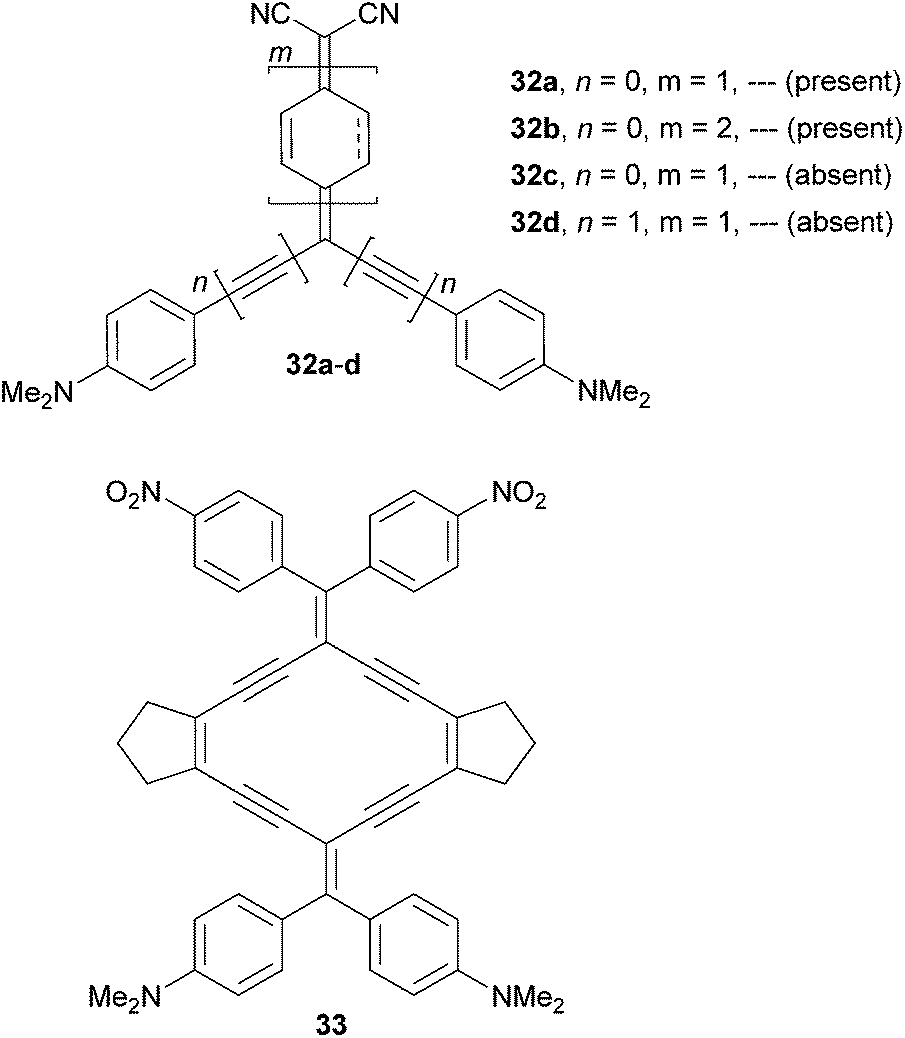 | ||
| Fig. 22 Proaromatic chromophores32a–d and radiaannulene 33. | ||
In contrast to 32a–b, molecules 32c–d were oxidized (quinoid) only partially. Further expansion of the benzoquinone ring has been demonstrated by the synthesis of the radiaannulene quinoid molecule 33. Evidence of proaromaticity has been presented in terms of structural, spectroscopic, electrochemical and computed data (Table 14). When comparing a pair of fully/partially quinoid chromophores 32a/32c the HOMO–LUMO gap decreased from 1.83 to 1.47 eV and the absorption maximum was red-shifted by 170 nm. Extension of the π-system by two additional acetylenic spacers (32c → 32d) had a diminished effect, whereas insertion of an additional benzoquinone ring as in 32b shifted the first oxidation potential to negative values; the HOMO–LUMO gap was reduced to 0.91 eV and the longest-wavelength absorption maximum reached the NIR region (845 nm). This series of molecules nicely demonstrates how powerful tuning through proaromatic spacers can be (ΔEg = 0.92 eV, Δλmax = 317 nm). Moreover, the observed negative solvatochromism further confirms the strong charge-separation in the ground state. Radiaannulene 33 showed electrochemical and optical properties at the level of chromophores 32a/32d.
Currently well-elaborated proaromatic electron donating groups comprise mostly 4H-pyran-4-ylidene,12 pyridine-4-ylidene71,72 and 1,3-dithiol-2-ylidene (dithiafulvene)73,74 (Fig. 23).
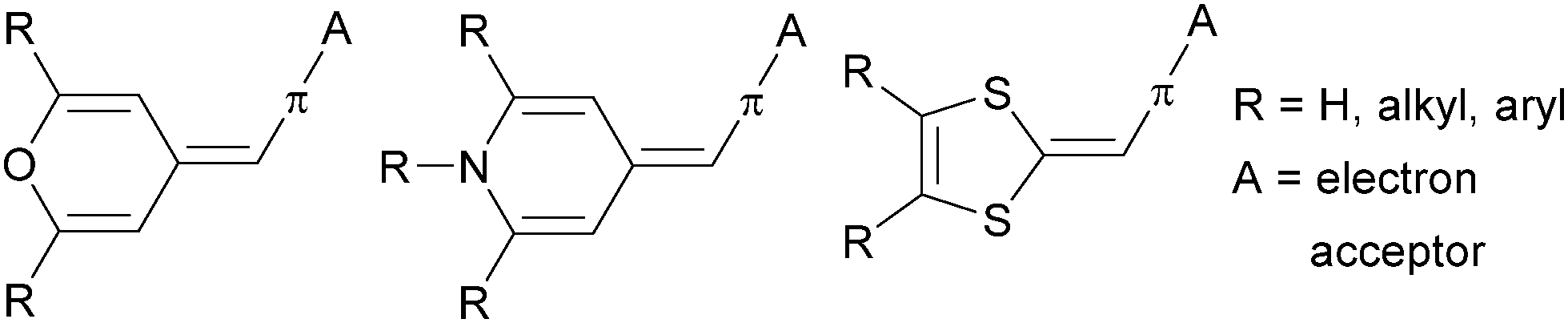 | ||
| Fig. 23 Proaromatic donor moieties. | ||
4H-Pyran-4-ylidene and its application as an electron donor has been extensively studied by Garín et al.12a,b For instance, they have studied push–pull molecules 34a–d (Fig. 24) end-capped with a proaromatic donor and tricyanofuran (TCF) acceptor with systematically extended polyenic central spacer. Later on, the pyran moiety has also been used as a central spacer and the linear chromophore arrangement as in 35a–b has been completed with the V-shaped molecule 36 (Fig. 24). The fundamental optoelectronic properties and optical nonlinearities of these molecules were further investigated (Table 15). Gradual extension of the polyenic central spacer in 34a–d reduced the HOMO–LUMO gap and red-shifted the λmax up to 688 nm. The NLO response (μβ0) increased dramatically from 90 (34a) to 3110 × 10−48 esu (34d). Similar trends can be seen in molecules 35a–b; however when going from 35a to 36 (linear vs. V-shaped arrangement), the second π-conjugated branch in 36 had a small influence on the optoelectronic properties (Table 15).
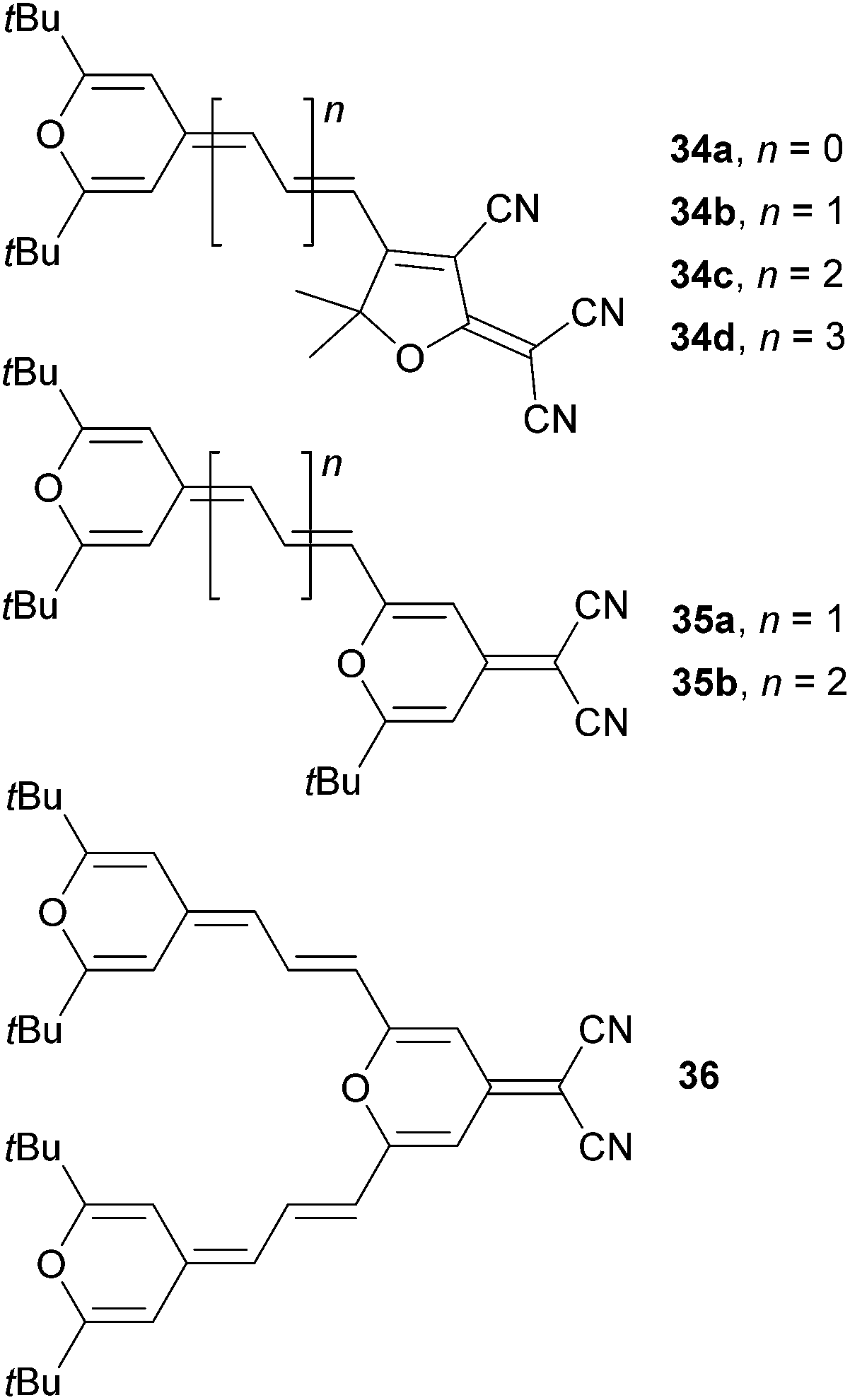 | ||
| Fig. 24 4H-Pyran-4-ylidene proaromatic donor. | ||
| Comp. | E ox,1 a [V] | E red,1 a [V] | E g [eV] | λ max b [nm (eV)] | μβ 0 c [10−48 esu] |
|---|---|---|---|---|---|
| a Measured in dichloromethane, vs. Ag/AgCl. b Measured in dichloromethane. c EFISH experiment at 1907 nm (dichloromethane). d μβ value. | |||||
| 34a | 1.32 | −1.10 | 2.42 | 513(2.42) | 90 |
| 34b | 1.00 | −0.83 | 1.83 | 592(2.09) | 310 |
| 34c | 0.68 | −0.77 | 1.45 | 675(1.84) | 1520 |
| 34d | 0.51 | −0.69 | 1.20 | 688(1.80) | 3110 |
| 35a | 0.76 | −1.18 | 1.94 | 587(2.11) | 652 |
| 35b | 0.59 | −1.21 | 1.80 | 602(2.06) | 1370 |
| 36 | 0.72 | −1.12 | 1.84 | 633(1.96) | 1500d |
Dipolar and quadrupolar organic dyes with a diphenylmethylenepyran donor were also studied by Achelle et al. (Fig. 25 and Table 16).12d Linear molecules 37a–d were synthesized by Wittig olefination, Sonogashira cross-coupling and eventually Knoevenagel condensation. Bismethylenepyran derivative 38 was prepared by oxidative dimerization of 37a in the presence of a I2/Na2S2O3 oxidation/reduction system. In contrast to the extension of the π-system as in 34a–d (Table 15), variation of the peripheral acceptor (CN → NO2 → CF3 → DCV) did not affect the electrochemically measured first oxidation and reduction potentials. This can be explained by the formation of a pyrylium radical 34+˙, which undergoes facile electrochemical dimerization to bispyrylium compounds similar to 382+. The longest wavelength absorption maxima of 37a–d and 38 shift slightly bathochromically with increased electron withdrawing ability of the appended A-groups. The highest NLO coefficient has been measured for DCV-substituted derivative 37d (μβ = 1150 × 10−48 esu); quadrupolar derivative 38 showed average NLO performance.
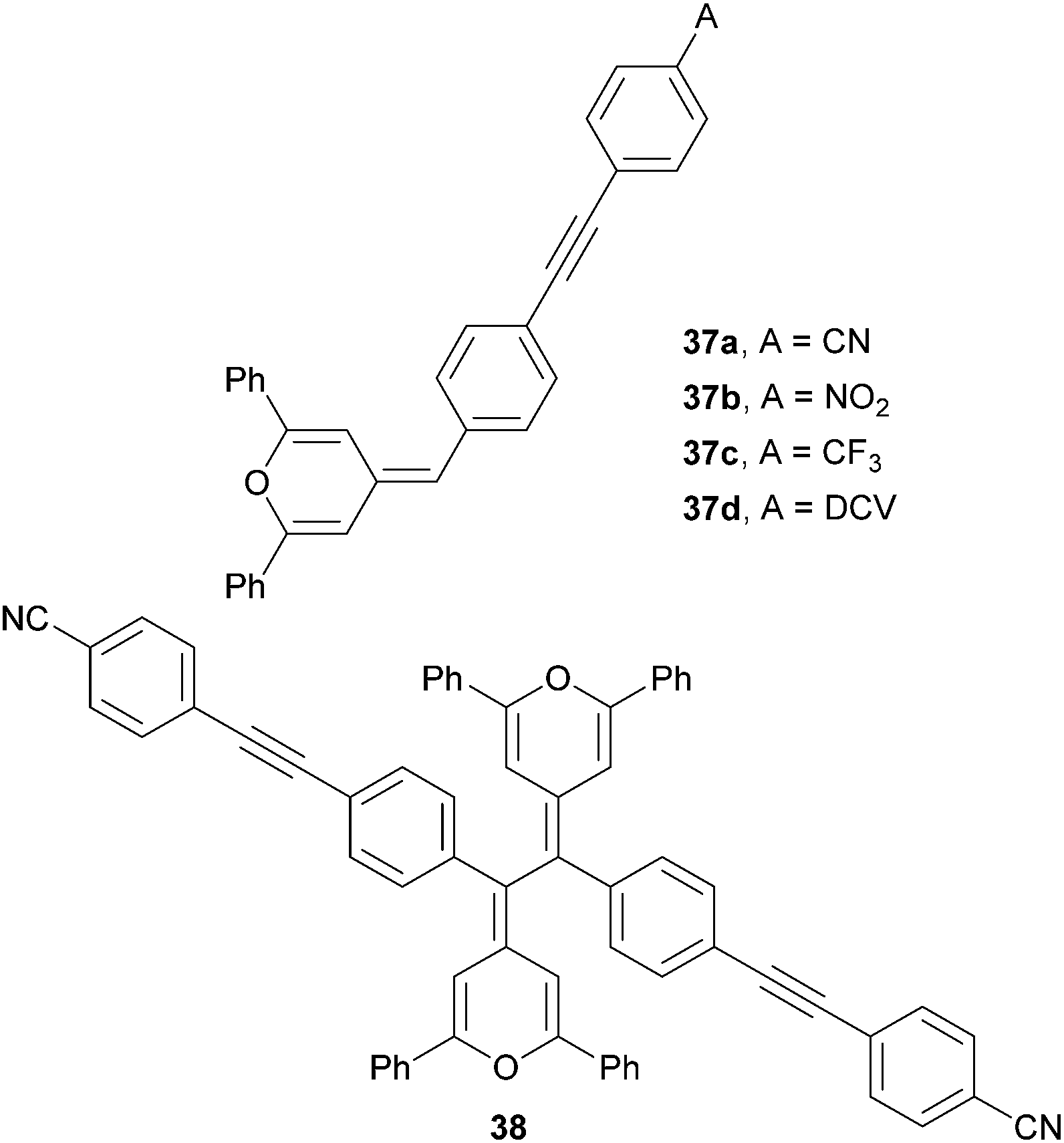 | ||
| Fig. 25 Dipolar and quadrupolar NLO phores bearing diphenylmethylenepyran donor(s). | ||
| Comp. | E ox,1 a [V] | E red,1 a [V] | λ max c [nm (eV)] | μβ d [10−48 esu] |
|---|---|---|---|---|
| a Measured in dichloromethane, vs. Fc+/Fc. b Represent reduction of the bispyrylium dication. c Measured in dichloromethane. d EFISH experiment at 1907 nm (chloroform). | ||||
| 37a | 0.31 | −0.87b | 414(3.00) | 350 |
| 37b | 0.32 | −0.86b | 432(2.87) | 200 |
| 37c | 0.32 | −0.89b | 403(3.08) | 700 |
| 37d | 0.31 | −0.85b | 463(2.68) | 1150 |
| 38 | −0.16 | — | 453sh(2.74) | 830 |
4-Methylene chalcogenapyrans as proaromatic donors were also utilized in the synthesis of push–pull Fischer type carbene complexes 39a–c (Fig. 26).12c Depending on the chalcogen atom involved, these molecules exhibited (non)linear optical properties with λmax = 594–613 nm and μβ = 1219–2348 × 10−48 esu, respectively. Thiopyran-based molecule 39b delivered the strongest NLO response.
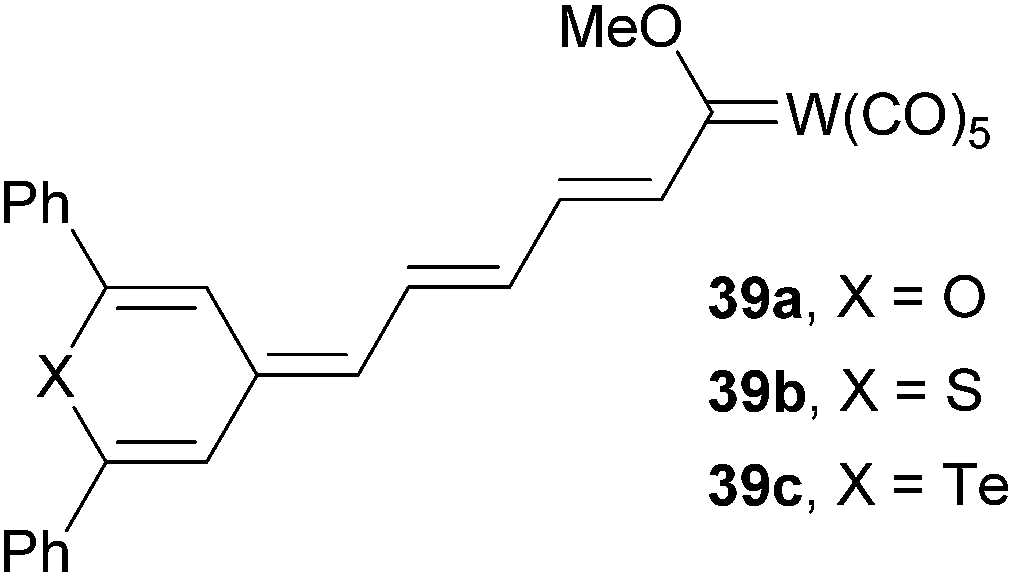 | ||
| Fig. 26 Chalogenapyrans in push–pull Fisher type carbene complexes. | ||
A series of molecules 40a–c was prepared by Woolhouse et al. (Fig. 27, Table 17).71 In these molecules, proaromatic pyridine-4-ylidene and TCF act as electron donor and acceptor, respectively. When comparing 40a–c with isolobal 34a–c (Fig. 24), the later series showed a more rapid bathochromic shift and a larger increase in the NLO coefficient, accompanied by the extension of the polyene spacer.
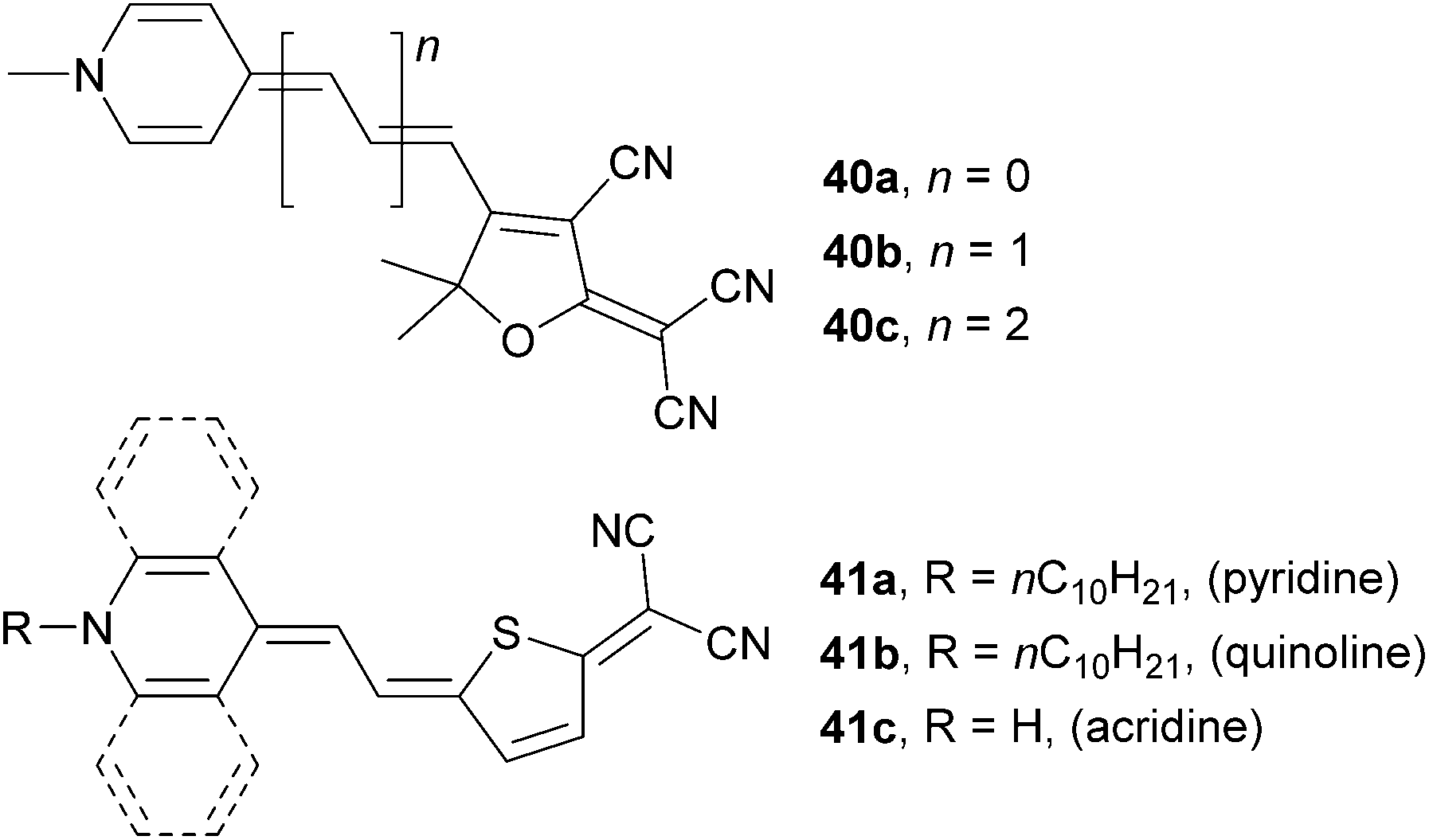 | ||
| Fig. 27 Pyridine-4-ylidene and related proaromatic donors. | ||
| Comp. | λ max [nm (eV)] | β 0 [10−30 esu] |
|---|---|---|
| a Measured in DMF. b HRS experiment in DMSO. c Measured in DMSO. d Deduced from the electro-optical absorption and UV/Vis spectra using a two-level model, in 10−50 CV−2 m3 (dioxane). | ||
| 40a | 570(2.17)a | 125b |
| 40b | 600(2.07)a | 360b |
| 40c | 615(2.02)a | 500b |
| 41a | 619(2.00)c | −92 ± 15d |
| 41b | 714(1.74)c | 66 ± 18d |
| 41c | 787(1.58)c | 338 ± 14d |
Abbotto et al. focused on the synthesis and thorough structure–property relationships study on molecules 41a–c bearing pyridine-4-ylidene donor gradually annulated with one or two benzene rings (quinoline and acridine derivatives, Fig. 27).72 As can be seen from the data given in Table 17, fusion of the benzene(s) to the pyridine-4-ylidine donor red-shifts the absorption maxima and the NLO coefficient to positive values. It was concluded that 41a–c are highly polarizable molecules, primarily zwitterionic and their NLO response strongly depends on the environment (solvent/matrix). In general, the NLO activity estimation should be carried out in various media to avoid unfortunate choice of solvent as dictated by instrumental availability and solubility properties.
Garín et al. have also investigated proaromatic dithiafulvene donor moiety attached either on 1,1,3-tricyano-2-phenylpropene (42) or TCF (43) acceptors (Fig. 28).73,74
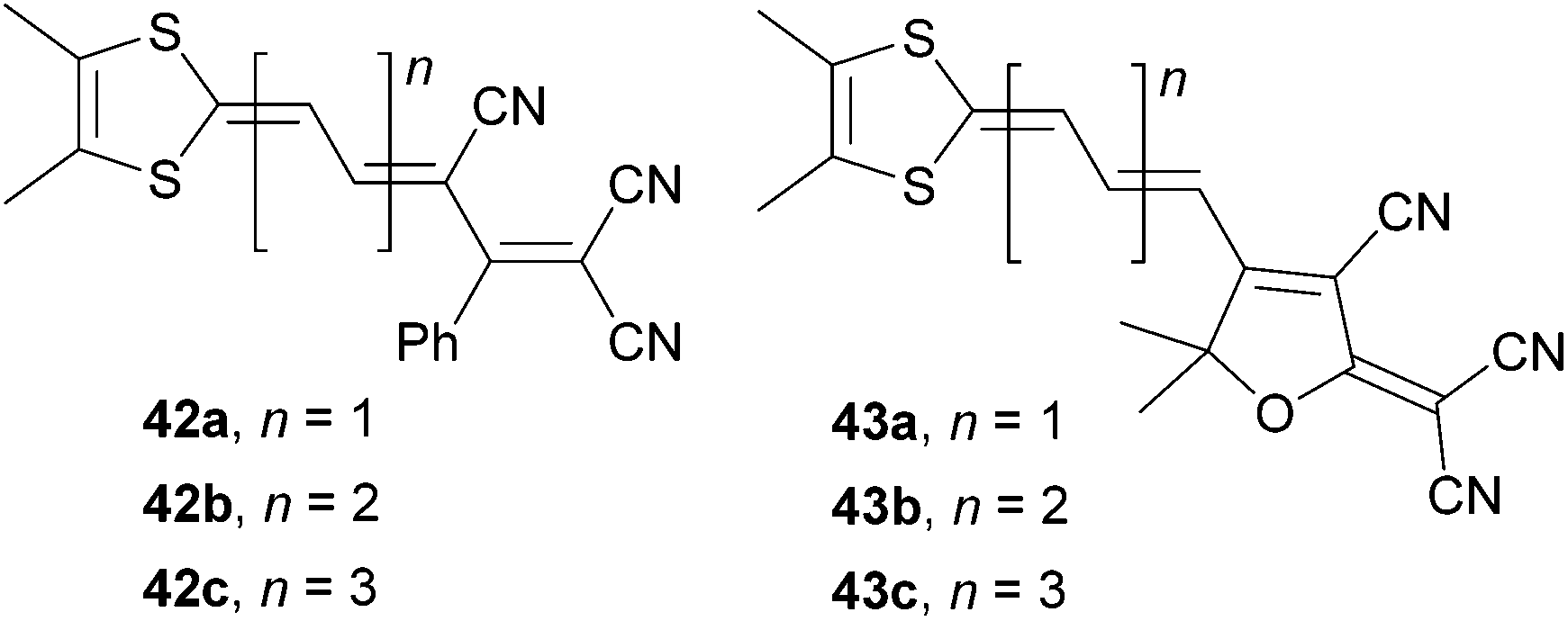 | ||
| Fig. 28 Dithiafulvene proaromatic donors in push–pull molecules. | ||
As expected, the HOMO–LUMO gap was narrowed, the λmax was shifted bathochromically and the NLO coefficient was raised with extension of the central olefinic backbone (Table 18). A comparison of molecules 34c (Eg = 1.45 eV, λmax = 1.84 eV and μβ0 = 1520 × 10−48 esu), 40c (λmax = 2.02 eV and μβ0 = 8900 × 10−48 esu) and 43b (Eg = 1.47 eV, λmax = 1.69 eV and μβ0 = 1938 × 10−48 esu) bearing TCF acceptor, the same polyenic spacer (n = 2) and differ only in the attached proaromatic electron donor seems to be reasonable at this point. Molecules 34c and 43b having 4H-pyran-4-ylidene and dithiafulvene donors showed very similar electrochemical and optical gaps. The optical gap of pyridine-4-ylidine-substituted chromophore 40c is higher by more than 0.2 eV but this molecule delivered 4–5 times stronger NLO response than analogues 34c and 43b. However, an artificial enrichment of this value due to the used calculated dipole moment μ cannot be excluded.
| Comp. | E ox,1 a [V] | E red,1 a [V] | E g [eV] | λ max b [nm (eV)] | μβ 0 c [10−48 esu] |
|---|---|---|---|---|---|
| a Measured in dichloromethane, vs. Ag/AgCl. b Measured in dichloromethane. c EFISH experiment at 1907 nm (dichloromethane). | |||||
| 42a | 1.25 | −0.85 | 2.10 | 590(2.10) | 135 |
| 42b | 0.85 | −0.68 | 1.53 | 688(1.80) | 935 |
| 42c | 0.68 | −0.57 | 1.25 | 715(1.73) | 3640 |
| 43a | 0.94 | −0.78 | 1.72 | 653(1.90) | 609 |
| 43b | 0.75 | −0.72 | 1.47 | 732(1.69) | 1938 |
| 43c | 0.61 | −0.64 | 1.25 | 835sh(1.49) | 6173 |
Anyway, very similar trends can be seen in the comparative study carried out by Garín et al.75 Three analogous molecules 44, 45 and 46 (Fig. 29) bearing the aforementioned proaromatic donors were synthesized and investigated. Despite its highest electrochemical and optical gaps, the NLO performance of pyridine-4-ylidene-substituted derivative 45 is 4 to 6-times higher than that of 44 and 46. Hence, pyridine-4-ylidenene proaromatic donor seems to be the most efficient one when designing new NLO phore with high nonlinearities.
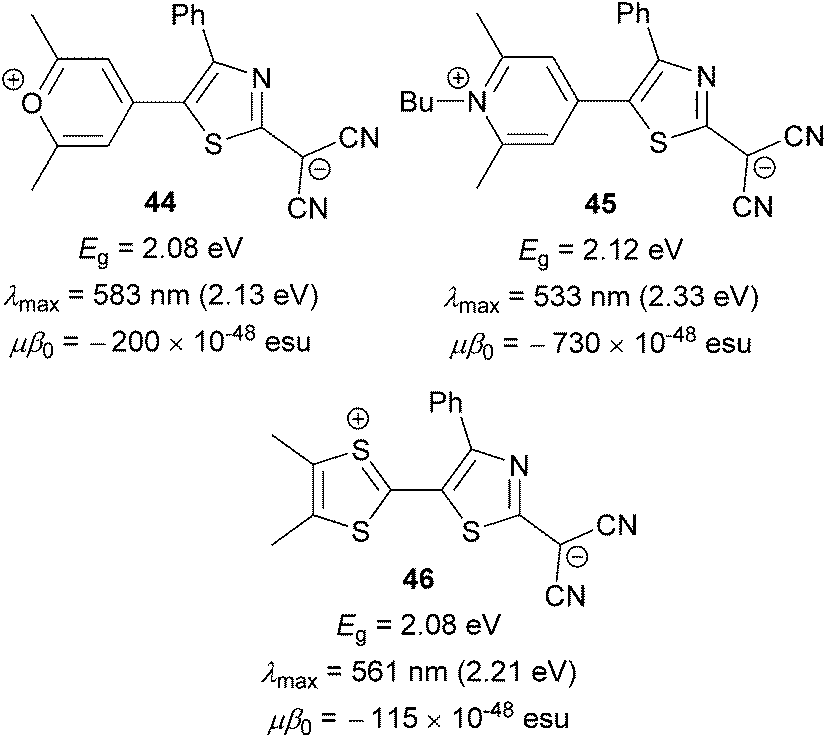 | ||
| Fig. 29 Performance of pyran-4-ylidene, pyridine-4-ylidene and dithiafulvene proaromatic donors in push–pull molecules. | ||
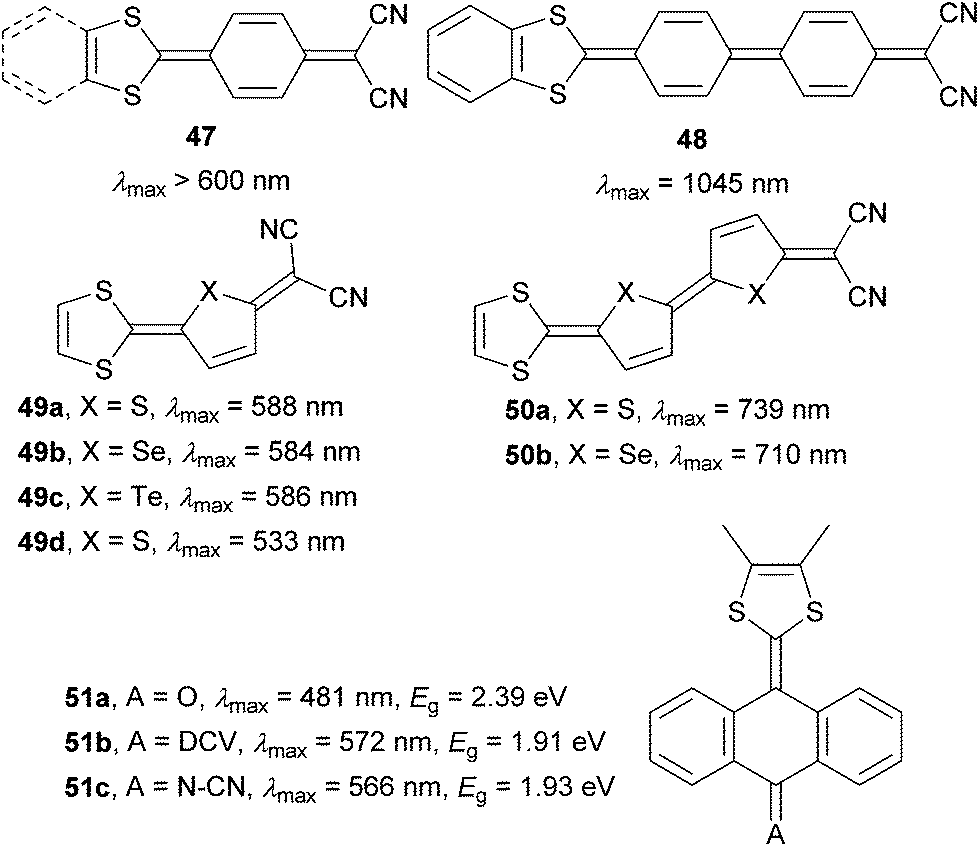
A combination of proaromatic donor and acceptor parts would render the most powerful D–Q–A chromophores. The first synthesis of such fully quinoid push–pull molecules 47 carried out by Gompper et al. can be dated back to 1966–1968 (Fig. 30).76 In late 90’s, extended molecules 48–51 were prepared and spectrally/electrochemically investigated by Otsubo77,78 and Bryce79 (Fig. 30). As can be seen, extension of the parent molecule 47 by an additional benzoquinone ring (48) shifted the absorption maxima by 400 nm. Likewise, whereas chalcogen exchange in 49 did not significantly affect the optical properties, extension of the π-system by one additional thienylene unit resulted in chromophores 50a–b with the absorption maxima exceeding 700 nm. Property tuning in anthraquinone-derived molecules 51a has been accomplished by attaching stronger DCV and N–CN acceptors. This provided chromophores 51b–c with narrowed band gaps and bathochromically shifted CT-bands.
 | ||
| Fig. 30 Fully quinoid dyes 47–51. | ||
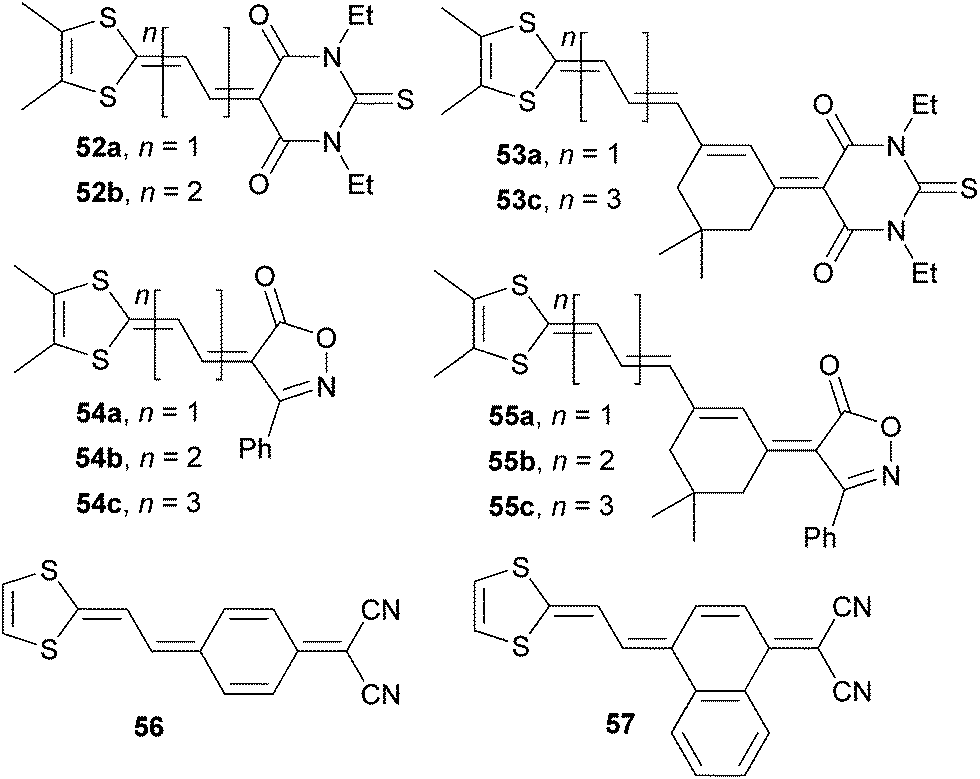
Novel doubly proaromatic NLO phores 52–57 were investigated by Garín et al. (Fig. 31).80–82 In molecules 52 and 53 is the 4,5-dimethyldithiafulvene donor linked via expanded olefinic bridge to proaromatic thiobarbituric acid. Extension of the π-system lowered the HOMO–LUMO gap, red-shifted the CT-band and increased the NLO coefficient μβ0 (compare pairs of chromophores 52a/52b or 53a/53c, Table 19). Although well polarizable, polyenic spacers often suffer from thermal, chemical and conformational instability. This is also the reason why molecule 53b was not isolated. However, a comparison of 52b and 53a possessing the same number of double bonds (π-electrons) and conformationally (un)locked π-system, reveals that the latter (isophorone-locked) features narrowed band gap and two-times higher NLO response. Very similar conclusions can also be evaluated from the series 54a–c and 55a–c in which the six-membered thiobarbituric acid acceptor was replaced by five-membered isoxazolone. A comparison across both series reveals similar electrochemical gaps, slightly hypsochromically shifted CT-bands for isoxazolone-substituted molecules 54 and 55 (an additional blue-shift observed for isophorone-bridged molecules 55a–c is also noticeable) and generally higher nonlinearities measured for isoxazolone derivatives 54 and 55 (compare for instance pair of chromophores 52b/54b and 53c/55c, Table 19). Effect of benzo-annulation was demonstrated on molecules 56 and 57. Likewise to observation on annulation at pyridine-4-ylidene donor made by Abbotto (41a–c, Fig. 27 and Table 17), chromophore 57 showed bathochromically shifted CT-band and positive value of μβ0 (Table 19). This further implies that NLO phore 56 possesses considerably zwitterionic (quinoid) ground state which was also evidenced by X-ray analysis and DFT calculations.
 | ||
| Fig. 31 Novel doubly proaromatic NLO phores. | ||
| Comp. | E ox,1 a [V] | E red,1 a [V] | E g [eV] | λ max b [nm (eV)] | μβ 0 c [10−48 esu] |
|---|---|---|---|---|---|
| a Measured in dichloromethane, vs. Ag/AgCl. b Measured in dichloromethane. c EFISH experiment at 1907 nm (dichloromethane). | |||||
| 52a | 1.16 | −1.00 | 2.16 | 558(2.22) | 60 |
| 52b | 0.87 | −0.85 | 1.72 | 642(1.93) | 525 |
| 53a | 0.65 | −0.81 | 1.46 | 638(1.94) | 1135 |
| 53c | 0.48 | −0.77 | 1.25 | 638(1.94) | 1350 |
| 54a | 1.06 | −1.12 | 2.18 | 547(2.27) | 153 |
| 54b | 0.77 | −0.93 | 1.70 | 621sh(2.00) | 565 |
| 54c | 0.59 | −0.82 | 1.41 | 633(1.96) | 1091 |
| 55a | 0.59 | −0.95 | 1.50 | 606(2.05) | 1475 |
| 55b | 0.50 | −0.91 | 1.41 | 614(2.02) | 2229 |
| 55c | 0.43 | −0.86 | 1.29 | 622(1.99) | 2843 |
| 56 | 0.63 | −0.64 | 1.27 | 652(1.90) | −667 |
| 57 | 0.74 | −0.63 | 1.37 | 672(1.85) | 115 |
6. Tuning through the acceptor
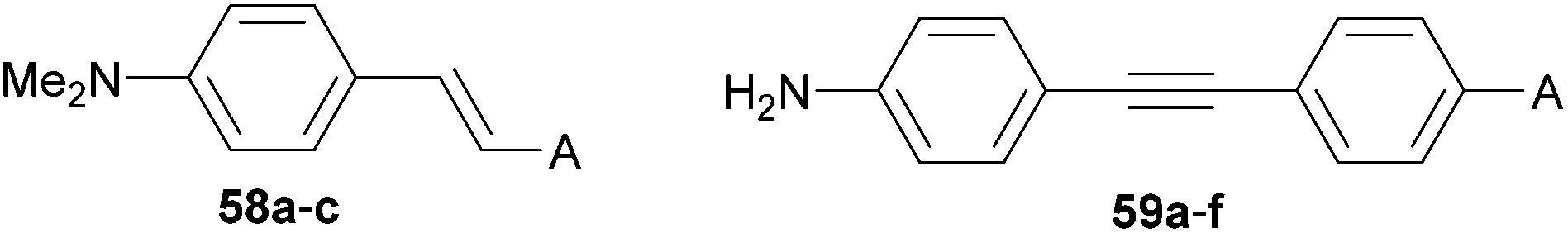
The property tuning in D–π–A molecules can also be accomplished via alternation, addition and position of the acceptor part. The A-part is most frequently represented by functional groups with negative resonance and inductive effects such as cyano, nitro and carbonyl as well as their combinations.For example, a variation of electron withdrawing groups on the amino-substituted styrenes 58a–c and diphenylacetylenes 59a–f has been performed by Cheng et al. (Fig. 32, Table 20).37a,39b From the data given in Table 20 we can deduce that nitro and cyano groups induce the most efficient ICT in the push–pull molecules and thus red-shift the absorption edge and increase the first-order hyperpolarizability. The formyl group also seems to be very powerful, however this moiety is chemically much more reactive and therefore less commonly used. Moderate polarizations/nonlinearities were observed for molecules 59a–59d bearing acyl, benzoyl methoxycarbonyl and methylsulfonyl groups.
 | ||
| Fig. 32 Acceptor variation on styrene and diphenylacetylene D–π–A systems. | ||
| Comp. | A | μ g/Δμa [10−18 esu] | λ max b [nm (eV)] | β/γc [10−30/36 esu] |
|---|---|---|---|---|
| a Ground state dipole moments and its change upon excitation (solvatochromism of the CT-band). b Measured in chloroform. c First- and second-order hyperpolarizabilities measured by EFISH and THG experiments (1910 nm). | ||||
| 58a | CN | 6.0/— | 364(3.41) | 23/29 |
| 58b | CHO | 5.6/— | 384(3.23) | 30/63 |
| 58c | NO2 | 6.5/— | 438(2.83) | 50/— |
| 59a | COCH3 | 3.3/12.6 | 336(3.69) | 12/— |
| 59b | COPh | 3.7/9.9 | 352(3.52) | 19/— |
| 59c | CO2Me | 3.8/10.6 | 337(3.68) | 15/— |
| 59d | SO2Me | 6.5/6.6 | 338(3.67) | 13/— |
| 59e | CN | 5.2/4.3 | 343(3.62) | 20/— |
| 59f | NO2 | 5.5/9.5 | 379(3.27) | 24/— |
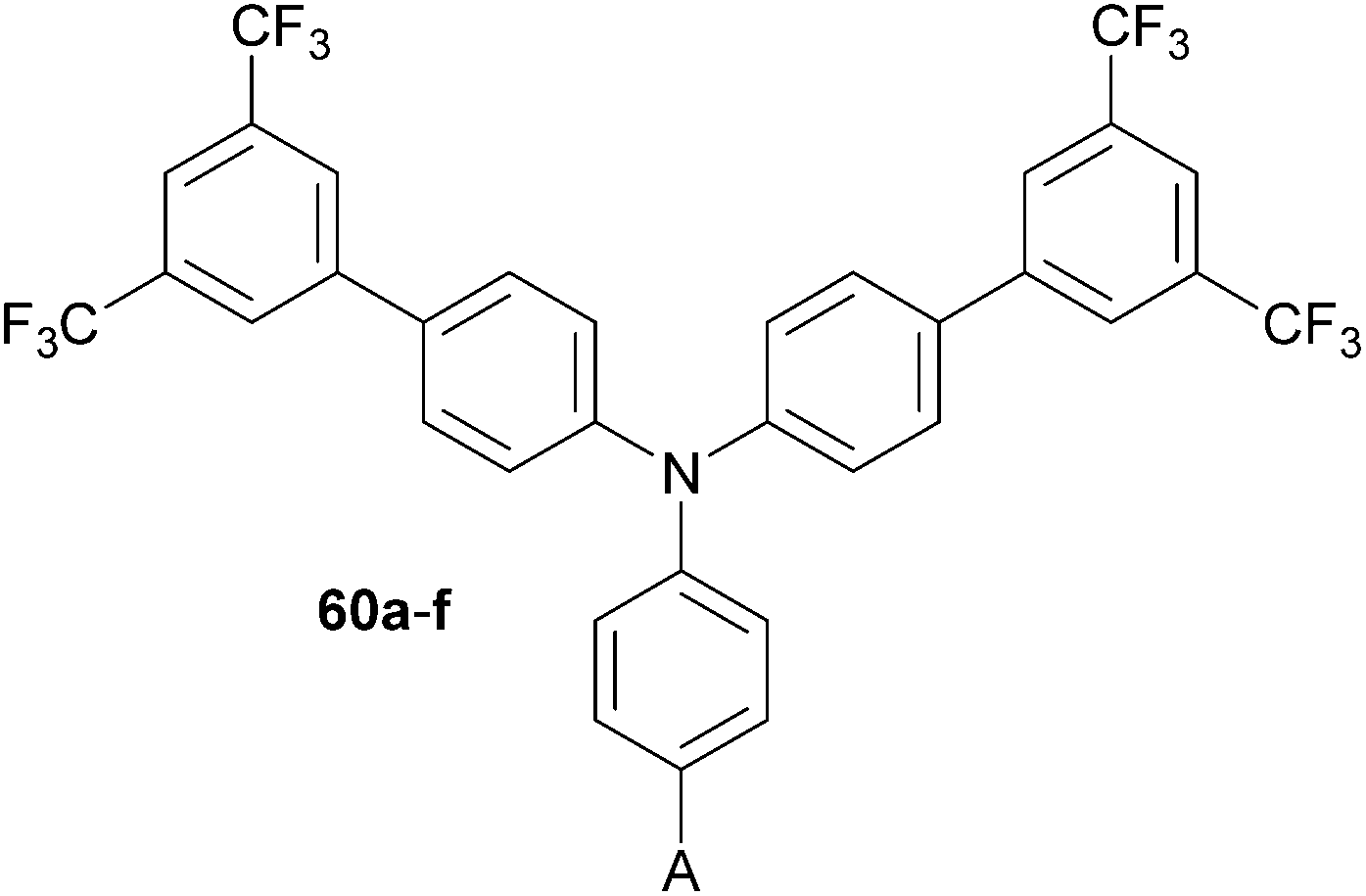
Vivas et al. carried out a comprehensive structure–property relationships study on non-centrosymmetric two-photon absorbing molecules 60a–f based on triarylamine (Fig. 33).83 The property tuning has been accomplished by variation of the acceptor group A appended at one of the aryl rings (Table 21).
 | ||
| Fig. 33 Various acceptors in non-centrosymmetric two-photon absorber 60. | ||
| Comp. | A | λ A/Emax a [nm] | Φ Emax | Δμc [D] | r eff/rd [%] | σ e [GM] |
|---|---|---|---|---|---|---|
| a Absorption/emission maxima (toluene). b vs. POPOP (Φ = 0.93 in cyclohexane). c Dipole moment changes upon excitation. d Degree of the ICT, where reff = Δμ/e defines the effective charge transfer (e = electron charge) and r is the distance between the amino core (donor) and the first atom of the acceptor (DFT calculation). e TPA cross-section measured by Z-scan at 775 nm in toluene [GM]. | ||||||
| 60a | H | 361/428 | 0.46b | 10.5 | 42 | 65 |
| 60b | CN | 347/422 | 0.24b | 9.5 | 34 | 58 |
| 60c | CHO | 359/437 | 0.19b | 9.0 | 35 | 48 |
| 60d | NO2 | 394/525 | 0.40b | 11.5 | 46 | 45 |
| 60e | CHC(CN)CO2Et |
426/510 | 0.13b | 13.0 | 54 | 115 |
| 60f | DCV | 439/535 | 0.24b | 13.5 | 54 | 125 |
On increasing the electron withdrawing capability of A (from 60b to 60f, Table 21), the absorption and emission maxima shift bathochromically, and the changes in dipole moment and degree of ICT increase from 9–13.5 D and 34 to 54%, respectively. As expected, the DCV acceptor causes the highest polarization. More importantly, the measured TPA cross-section mimics similar trends with the highest TPA coefficient σ = 125 GM measured for 60f.
The aforementioned examples imply that replacing simple electron withdrawing groups (e.g. CN or NO2) with more structurally complex electron accepting moieties (e.g. DCV) leads to push–pull molecules with significantly enhanced ICT. In this respect, various (hetero)aromates were utilized, not just as spacers and auxiliary donors (see above), but also as powerful electron acceptors. Several examples of property tuning through the acceptor are shown in Fig. 34.
 | ||
| Fig. 34 Property tuning through the acceptor (DMA = 4-(N,N-dimethylaminophenyl), DPA = 4-(N,N-diphenylamino)phenyl, PIT = 5-(piperidin-1-yl)thiophene-2-yl). | ||
Dimethylanilino-substituted D–π–A fluorescent thiophenes 61a–c were prepared and investigated by Blanchard-Desce and Raposo.40b,84 Gradual replacement of the formyl group in 61a by DCV (61b) and N,N′-diethylthiobarbituric acid (61c) acceptors led to bathochromic shift of the longest-wavelength absorption maxima and increased two-photon absorption cross-section, as well as higher thermal stability (Table 22). Moylan et al. carried out property tuning through the acceptor in a series of azo NLO phores 62a–c (Fig. 34).49 The aromatic 4-nitrophenyl acceptor (62a) has been replaced by heteroaromatic 5-nitrothiazole (62b) and 6-nitrobenzo[d]thiazole (62c). These structural changes resulted in chromophores with red-shifted λmax, higher ground-state dipole moments and first-order hyperpolarizabilities β0 within the range 57–71 × 10−30 esu. Thermal stability of 62a–c up to/over 300 °C is also noticeable. Thiobarbituric acid, although considered to be a very strong electron acceptor, can be replaced by the even stronger 3-dicyanomethylene-2,3-dihydrobenzo[d]thiophene-1,1-dioxide as in 63a–b (Fig. 34).40a Both chromophores showed the same ground-state dipole moments and similar changes upon excitation. However, with the same donor and π-linker as in 63a, 63b features a bathochromically shifted CT-band by 106 nm and 1.3-times higher nonlinearity. An interesting comparison of electron acceptors involving benzo[d]furan-3-one (64a), dicyanovinyl (DCV, 64b), N,N′-diethylthiobarbituric acid (64c) and 3-(dicyanomethylene)indane-1-one (64d) and their impact on (non)linear optical properties has been demonstrated by Roncali et al. (Fig. 34).85 The rigidified bithiophene central spacer end-capped with a DMA donor and gradual replacement of acceptors allowed optical tuning by 176 nm and increased the NLO response by almost 4-times. Hence, 3-(dicyanomethylene)indane-1-one proved to be the strongest donor in this series.
| Comp. | λ A/Emax [nm] | μ/Δμ [D] | NLO | T d [°C] |
|---|---|---|---|---|
| a Measured in chloroform. b TPA cross-section (wavelength) measured by two-photon excited fluorescence (TPEF) in chloroform, relative to fluorescein [GM]. c Melting point. d First-order hyperpolarizabilities measured by EFISH experiments (1907 nm) in chloroform. e Measured in dichloromethane. f μβ 0 product measured by EFISH experiments (1907 nm) in chloroform. | ||||
| 61a | 408/507a | −/− | 124(820)b | 190c |
| 61b | 512/609a | −/− | 176(1050)b | 209c |
| 61c | 572/669a | −/− | 428(1120)b | 260c |
| 62a | 486a/— | 5.9/— | 54.3d | 393 |
| 62b | 582a/— | 6.9/— | 68.2d | 295 |
| 62c | 550a/— | 7.2/— | 71.8d | 356 |
| 63a | 619a/— | 10.4/6.1 | 55d | 240 |
| 63b | 725a/— | 10.4/7.7 | 72d | 268 |
| 64a | 592e/— | — | 1500f | 300 |
| 64b | 614e/— | — | 1600f | 308 |
| 64c | 682e/— | — | 4400f | 235 |
| 64d | 768e/— | — | 5680f | 245 |
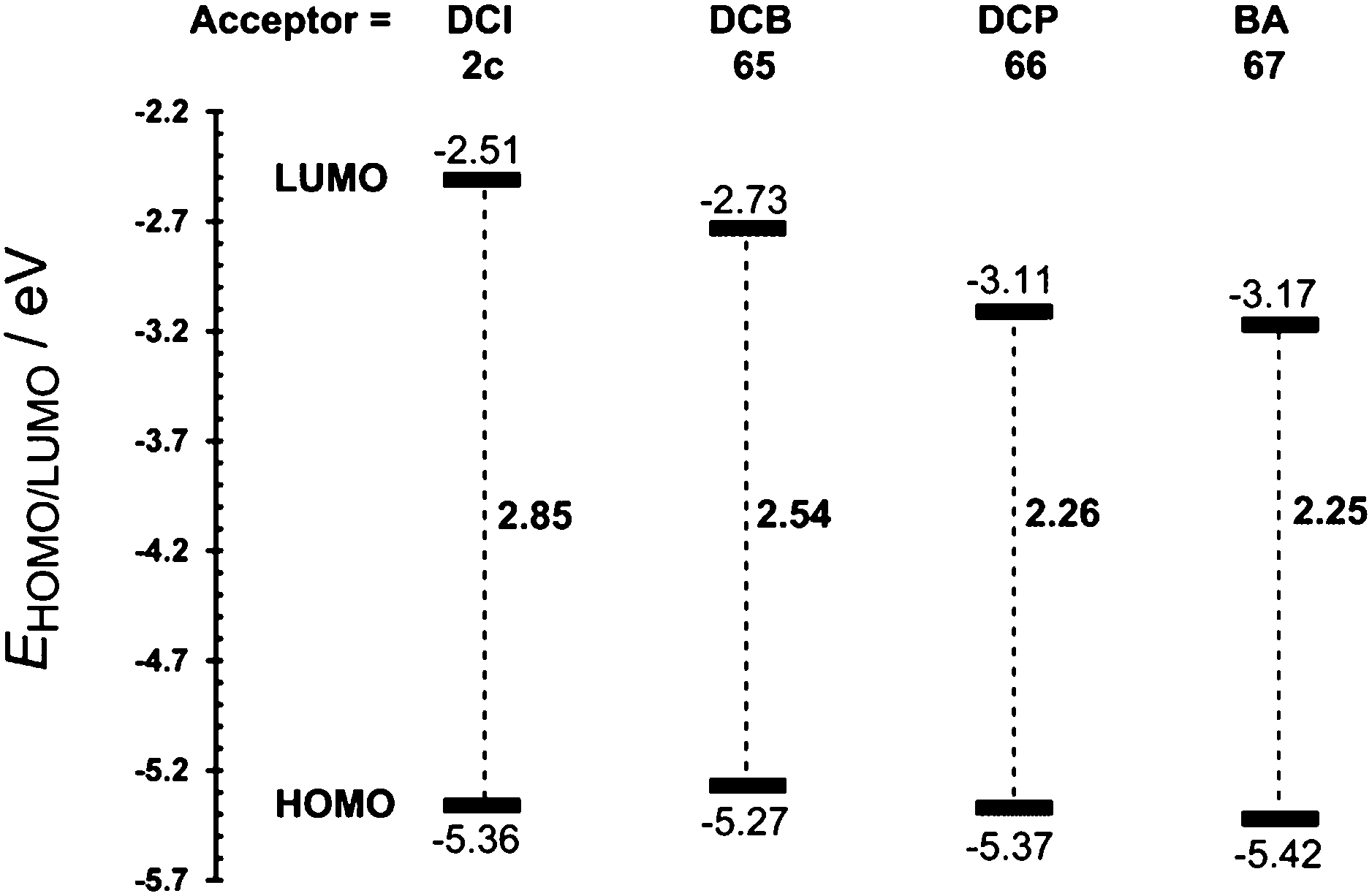
Based on our recent contributions, dicyano-substituted (hetero)aromatic acceptor units based on imidazole, benzene and pyrazine can be compared with barbituric acid here. Y-shaped dicyanoimidazole (DCI),32 X-shaped dicyanobenzene (DCB)86 and dicyanopyrazine (DCP),23 as well as N,N′-dibutylbarbituric acid (BA)87 derivatives 2c and 65–67 saturated by one or two DMA donor(s), can be chosen as model push–pull molecules (Fig. 35). Table 23 and the energy level diagram in Fig. 36 summarize the electrochemical and optical data as well as the measured NLO properties. As expected, the main changes in frontier orbitals are observed in the LUMO while the HOMO remained almost unaltered (EHOMO = 5.27–5.42 eV). When going from five-membered DCI (2c) to six-membered DCB (65) and DCP (66) acceptors, the HOMO–LUMO gap decreases stepwise by roughly 0.3 eV. The lowest gap was measured for DCP and BA derived compounds 66 and 67. Thus, DCI seems to be the weakest electron acceptor (two cyano groups attached at electron excessive imidazole), DCB behaves as a “standard aromatic acceptor” while replacement of two CH groups by nitrogens as in DCP narrowed the gap by an additional 0.3 eV (two cyano groups attached at the electron poor pyrazine).
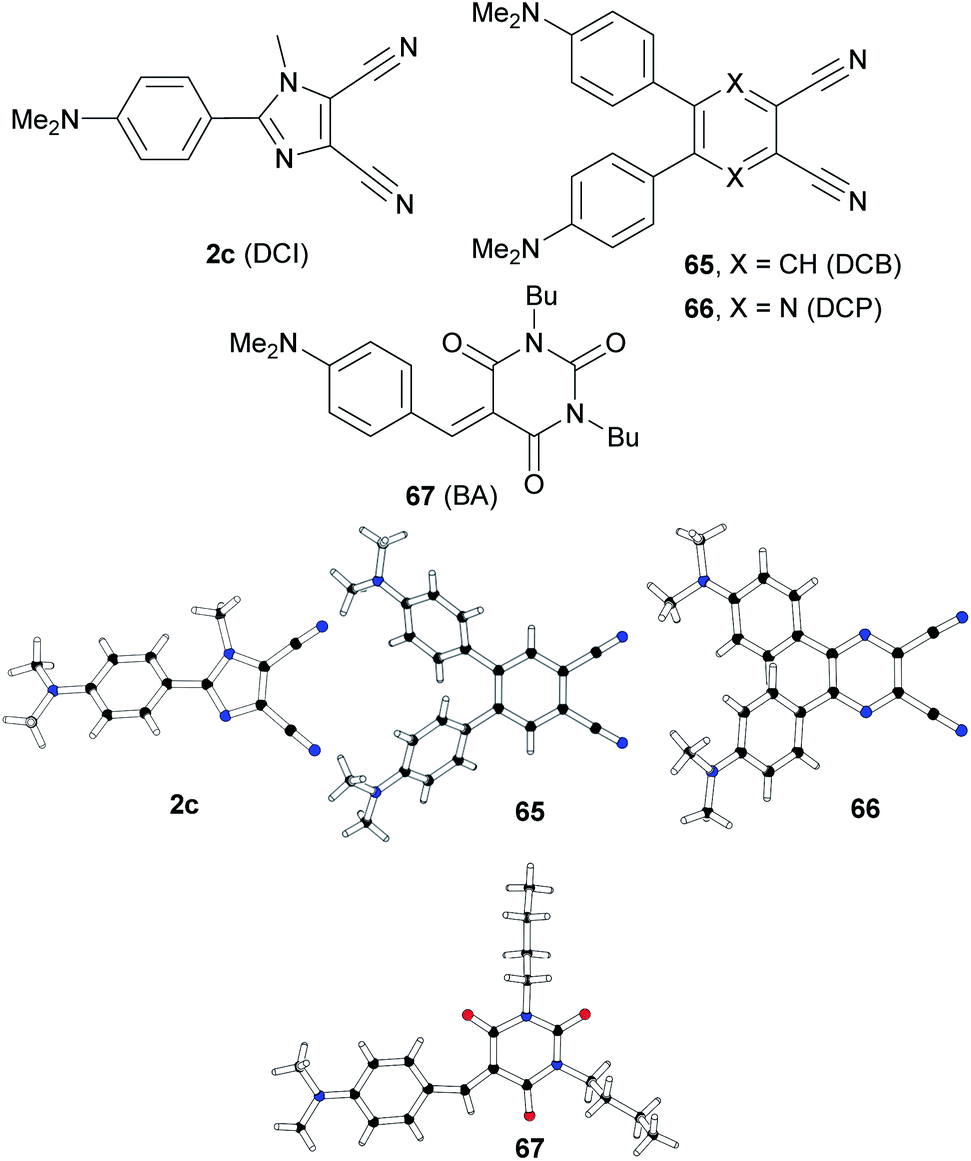 | ||
| Fig. 35 Structure of DCI, DCB, DCP and BA acceptor units in push–pull molecules 2c and 65–67, including their X-ray analyses. | ||
| Comp. | E HOMO a [eV] | E LUMO a [eV] | E g [eV] | λ max b [nm (eV)] | NLO |
|---|---|---|---|---|---|
| a Measured by CV, RDV and DC polarography in acetonitrile/DMF and recalculated.34 b Measured in dichloromethane at c = 10−5 M. c First order hyperpolarizability measured by HRS experiment (1064 nm) in dichloromethane (in 10−30 esu). d Second-order susceptibility of 65/66 incorporated into oligoetheracrylate photopolymer (1064 nm). The signal for 66 was below noise level (in pm V−1). e μβ product (β) measured by EFISH experiment (1907 nm) in chloroform (in 10−48(30) esu). | |||||
| 2c | −5.36 | −2.51 | 2.85 | 316(3.92) | 19.2c |
| 65 | −5.27 | −2.73 | 2.54 | 410(3.02) | 0.53d |
| 66 | −5.37 | −3.11 | 2.26 | 471(2.63) | —d |
| 67 | −5.42 | −3.17 | 2.25 | 462(2.68) | 270(36)e |
 | ||
| Fig. 36 Energy level diagram showing HOMO/LUMO of 2c and 65–67. | ||
One has to take into consideration that DCI is saturated by one DMA donor, whereas DCB and DCP are saturated by two DMA donors. However, the BA acceptor saturated by one DMA donor provided a similar gap (Eg = 2.25 eV) to DCP (Eg = 2.26 eV). It should also be mentioned that the DMA units in 2c, 65 and 66 are connected directly to the DCI, DCB and DCP acceptors with torsion angles 25–42° (see X-ray analyses in Fig. 35), whereas an additional methylene bridge in 67 causes planarization of the π-system (torsion angle about 4°). A significant twist of DMA(s) in 2c, 65 and 66 is most likely caused by the steric repulsion of the ortho-hydrogens and represents an additional barrier to efficient ICT.
The UV/Vis absorption spectra of 2c and 65–67 are shown in Fig. 37; the longest-wavelength absorption maxima are presented in Table 23. The optical properties mimic the trends seen by electrochemical measurements (Eg) with Δλmax of 155 nm. DCP- and BA-derived molecules 66 and 67 showed the most red-shifted CT-bands. Moreover, two particular CT-bands were observed for X-shaped molecule 66 and less developed also for 65. These bands are most likely originated by multiple interactions of each peripheral donor and the particular CN acceptor (Fig. 37). DCP-derivatives also proved to be very efficient and tuneable photoredox catalysts.23b
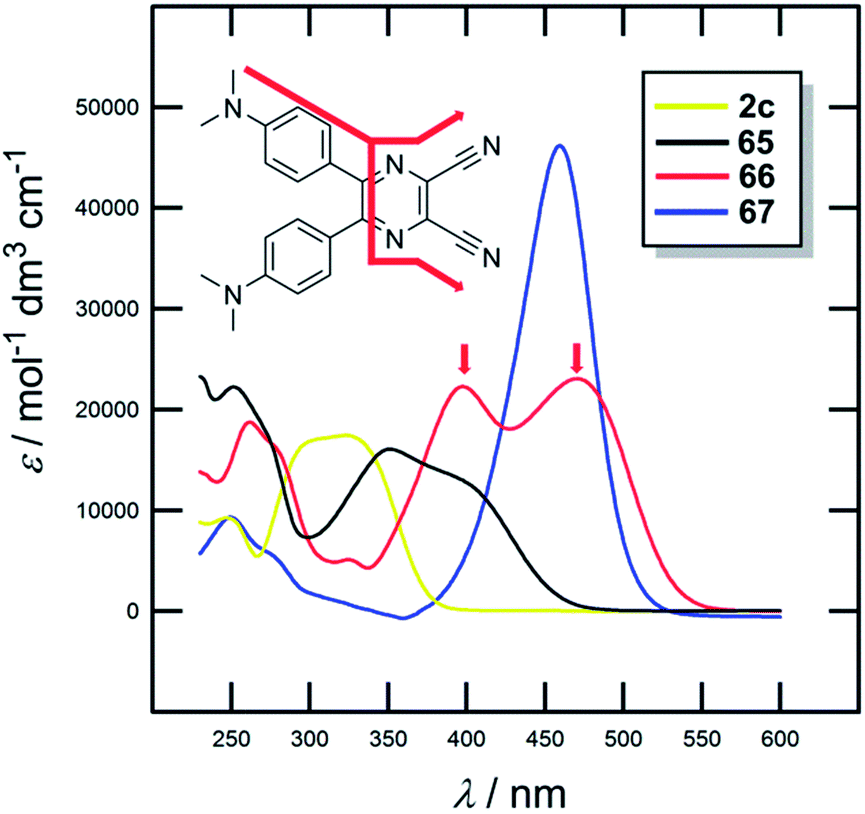 | ||
| Fig. 37 UV/Vis absorption spectra of 2c and 65–67. | ||
In principle, the withdrawing ability of azo-heterocyclic acceptors can further be improved by quaternization (see for instance molecules 12a–e, Fig. 7)41 or chelation by a (transition) metal.88 Electronic performance of the most frequently used cationic acceptor, pyridinium, can be for instance compared with fused five-membered benzo[d]thiazolium, as shown by Coe et al. (Fig. 38).89 Replacing the pyridinium acceptor in chromophoric cations 68 led to 69 with reduced HOMO–LUMO gap, red-shifted CT-band and pronounced NLO response measured by HRS at two laser frequencies (Table 24). Hence, the benzothiazolium ion can be considered as a stronger electron acceptor than the pyridinium ion. Further property tuning in 68–69 has been achieved by extension of the central olefinic spacer, which could also be substituted by thienylene units, as demonstrated by Clays et al.90
 | ||
| Fig. 38 Cationic chromophores. | ||
| Comp. | E ox,1 a [eV] | E red,1 a [V] | E g [eV] | λ max b [nm (eV)] | β 0 c [10−30 esu] |
|---|---|---|---|---|---|
| a Measured by CV in acetonitrile vs. Ag/AgCl. b Measured in acetonitrile. c Static first order hyperpolarizability β0 measured by HRS experiment (1300/800 nm lasers) in acetonitrile. | |||||
| 68 | 0.72 | −0.94 | 1.66 | 500(2.48) | 310/370 |
| 69 | 0.71 | −0.59 | 1.30 | 576(2.15) | 280/570 |
Barbituric acid related push–pull derivatives 70–73 were targeted in a brief contribution of Ikeda et al. (Fig. 39).91 In these molecules, the DMA donor was linked to N-unsubstituted barbituric acid (70), thiobarbituric acid (71), hydantoin (72) and rhodanine (73). When going from barbituric acid to thiobarbituric acid acceptors (70 → 71), the measured linear and nonlinear optical properties (λmax and μβ0) were shifted by 30 nm and 520 × 10−48 esu, respectively. This enhancement was attributed to the chalcogen effect. On the other hand, contraction of the six-membered BA derivative 70 to the hydantoin derivative 72 resulted in considerable hypsochromic shift and diminished NLO response. Compared to 72, two sulphur atoms in rhodanine derivative 73 shifted the CT-band by 90 nm and renewed the large NLO response (μβ0 = 1050 × 10−48 esu). Hence, the ring size and heteroatoms attached on it may play a very crucial role in the overall performance of the given heterocyclic acceptor.
 | ||
| Fig. 39 Barbituric acid and related acceptors (all data were measured in DMSO (EFISH experiment at 1060 nm)). | ||
Garín et al. have demonstrated property tuning through acceptors on push–pull molecules 74–78 (Fig. 40 and Table 25) comprising a proaromatic pyran-4-ylidene donor and complex acceptor groups such as thiobarbituric acid (74), isoxazolone (75), phenyl- and amino-substituted tricyanopropene (76 and 77) and tricyanofuran (78).12a The lowest electrochemical gaps were recorded for tricyano-substituted derivatives 76 and 78, which also reflects their most bathochromically shifted CT-bands, highest calculated dipole moments and their changes upon excitation, as well as the largest NLO response. In this respect, the TCF acceptor imbues the π-conjugated backbone with the strongest electronic effects and, in combination with the proaromatic pyran-4-ylidene donor, significantly polarizes the whole molecule. This A/D combination yields the very efficient NLO phore 78 with a μβ0 value of 1520 × 10−48 esu.
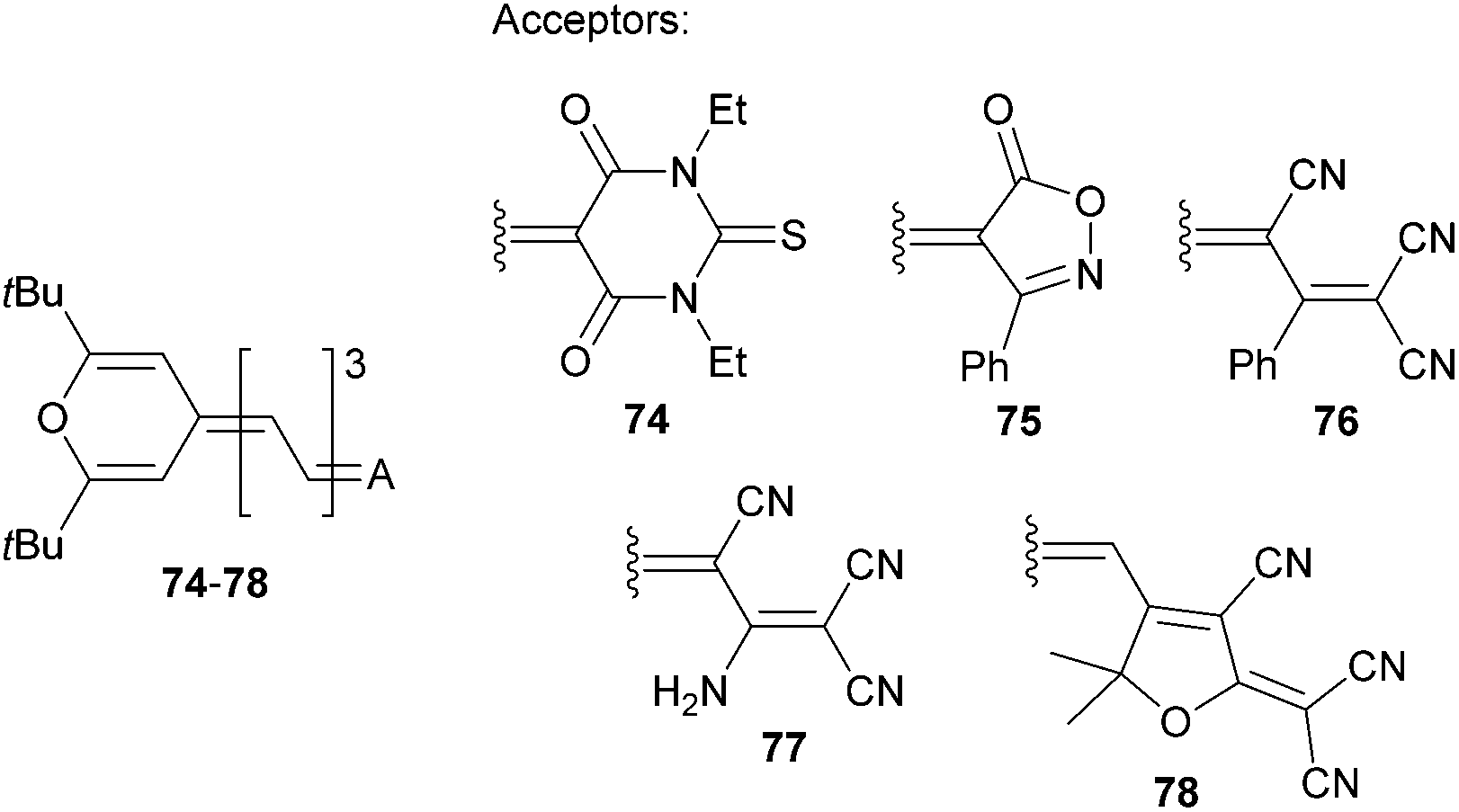 | ||
| Fig. 40 Acceptor variation in push–pull molecules 74–78 bearing the proaromatic pyran-4-ylidene donor. | ||
| Comp. | E ox,1 a [V] | E red,1 a [V] | E g [eV] | λ max b [nm (eV)] | μ/Δμc [D] | μβ 0 d [10−48 esu] |
|---|---|---|---|---|---|---|
| a Measured by CV in dichloromethane vs. Ag/AgCl. b Measured in dichloromethane. c Ground-state dipole moment and its change upon excitation (DFT calculation at the B3P86/6-31 G* level). d Measured by EFISH experiment (1907 nm) in dichloromethane. | ||||||
| 74 | 0.83 | −0.94 | 1.77 | 628(1.97) | 11.4/2.2 | 300 |
| 75 | 0.77 | −1.04 | 1.81 | 623(1.99) | 10.8/2.4 | 380 |
| 76 | 0.88 | −0.76 | 1.64 | 678(1.83) | 16.4/5.6 | 670 |
| 77 | 0.87 | −1.04 | 1.91 | 625(1.98) | 16.1/6.6 | 360 |
| 78 | 0.68 | −0.77 | 1.45 | 740(1.68) | 17.8/7.1 | 1520 |
The powerful accepting behavior of the TCF acceptor has further been investigated by Robinson et al.92 They developed molecules 79 and 80 (Fig. 41), which have a ferrocene donor connected via a thienylenevinylene bridge to the TCF and CF3PhTCF acceptors. The electron withdrawing properties of the latter one were improved by attaching nonconjugated CF3 and Ph groups. This relatively small but subtle structural change resulted in a significant red-shift of both high/low-energy (HE/LE) CT-bands and 1.2 times higher nonlinearity measured for 80.
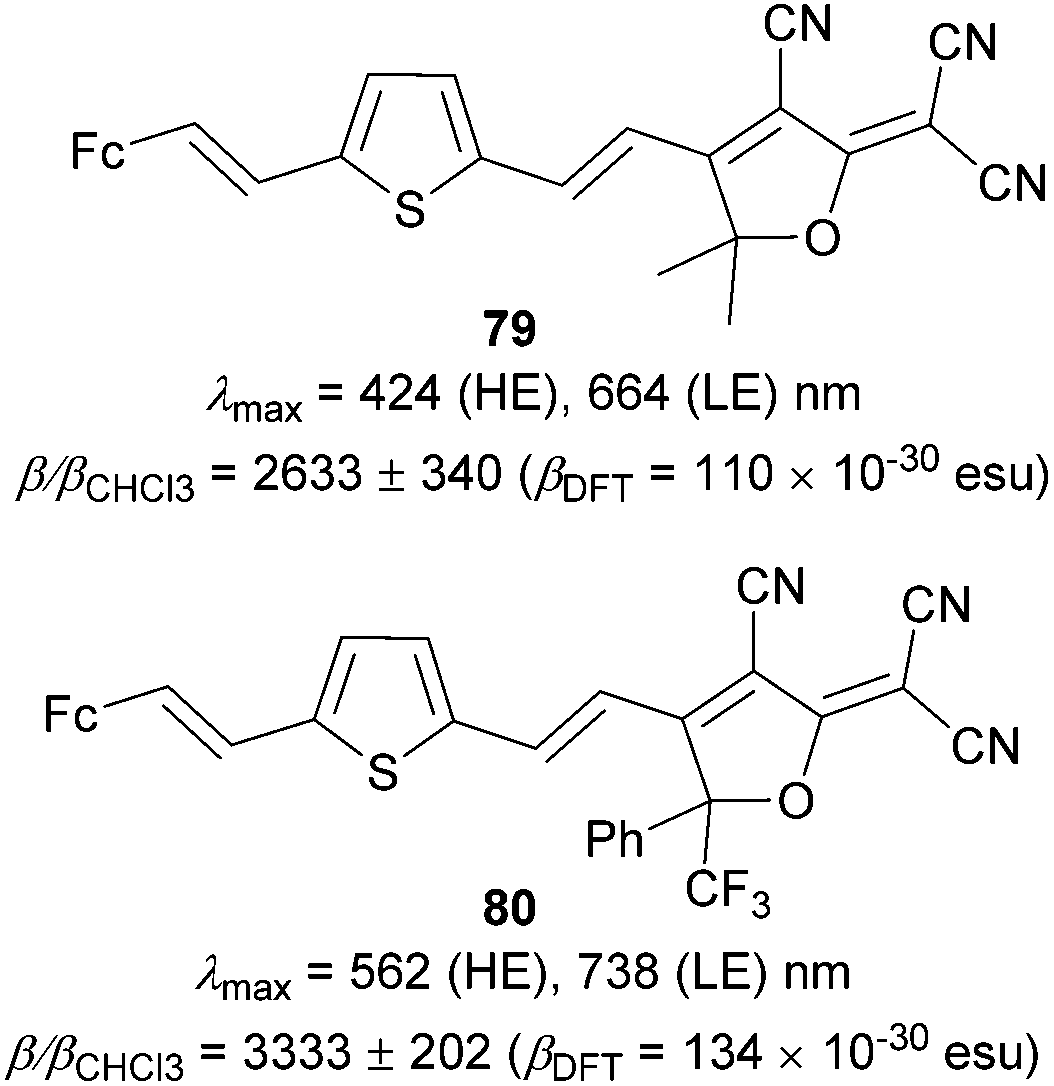 | ||
| Fig. 41 Modification of TCF acceptor (all data were measured in chloroform (HRS experiment at 1000 nm). | ||
Acceptor variation as a powerful tool for tailoring the properties of simple thiophene-based chromophores 81–90 (Fig. 42), with prospective applications ranging from photorefractive (PR) materials to BHJ solar cells, has been shown by Würthner et al.93,94
 | ||
| Fig. 42 Acceptor variation in push–pull molecules for PR materials and BHJ solar cells. | ||
Molecules 81–83, 85–86 and 90 were applied as active materials in solution-processed BHJ solar cells, while the remaining ones were tested as PR materials (Table 26). In a series of indane-1,3-dione derived chromophores 81–83, the electrochemical gap decreases steadily from 2.39 to 1.87 eV with increasing withdrawing ability of the acceptor moiety. This observation is also consistent with their CT-bands being red-shifted by 168 nm. Five- and six-membered heterocyclic moieties in 85–86 and 90 showed electrochemical gaps and positions of the longest-wavelength absorption maxima in a similar range as 82–83. However, their ground-state dipoles are twice as large, whereas the change upon excitation is diminished. This implies that, in contrast to indane-1,3-dione acceptors in 81–83, the structure of heterocyclic moieties in 85–86 and 90 (thiazole and tetrahydropyridine-2,6-dione derived) is shifted towards a zwitterionic character. OPVC devices based on these molecules and the PC61BM acceptor showed FF and power conversion efficiencies within the range 0.27–0.41 and 0.5–2.3%, respectively, with the highest performance measured for 82 bearing a 3-(dicyanomethylene)indan-1-one acceptor. Further optimization studies using this compound and different acceptors resulted in a device with power conversion efficiencies up to 4.5%.
| Comp. | E HOMO a [eV] | E LUMO a [eV] | E g [eV] | λ max [nm (eV)] | μ/Δμb [D] | F 0 c | FF/ηd [%] |
|---|---|---|---|---|---|---|---|
| a Measured by CV in dichloromethane vs. Fc/Fc+. b Ground-state dipole moment and its change upon excitation (EOAM in dioxane). c PR figure-of-merit in 10−74 m4 C2 mol V−2 kg−1. d Fill factor and power conversion efficiency. e Measured in thin film of the blend cast from chlorobenzene. f Measured in dichloromethane. | |||||||
| 81 | −5.56 | −3.17 | 2.39 | 532(2.33)e | 5.7/4.2 | 0.24 | 0.27/0.5 |
| 82 | −5.70 | −3.56 | 2.14 | 595(2.08)e | 8.6/4.1 | — | 0.41/2.3 |
| 83 | −5.61 | −3.74 | 1.87 | 700(1.77)e | 6.1/4.6 | — | 0.36/1.0 |
| 84 | — | — | — | 513(2.42)f | 6.7/7.2 | 0.32 | — |
| 85 | −5.45 | −3.55 | 1.90 | 689(1.80)e | 12.1/2.3 | — | 0.27/0.5 |
| 86 | −5.52 | −3.62 | 1.90 | 682(1.82)e | 13.1/2.5 | — | 0.35/1.1 |
| 87 | — | — | — | 500(2.48)f | 7.9/2.4 | 0.35 | — |
| 88 | — | — | — | 489(2.54)f | 7.0/2.1 | 0.26 | — |
| 89 | — | — | — | 647(1.92)f | 11.2/7.8 | 1.44 | — |
| 90 | −5.65 | −3.36 | 2.29 | 556(2.23)e | 12.6/1.7 | — | 0.32/0.9 |
Chromophores 81, 84 and 87–89 with indane-1,3-dione, thioxothiazolidinone, isoxazolone, pyrazolidinedione and thiophene derived acceptor units showed CT-bands around 500 nm (except for 89) and generally lower ground-state dipole moments as compared to 85–86. Whilst the photorefractive figures-of-merit F0 of 81, 84 and 87–88 are relatively low (∼0.3), the thiophene based chromophore 89 showed a considerably higher dipole moment as well as F0 = 1.44. Moreover, its narrow band absorption represents another significant advantage over the currently available PR chromophores and could lead to applications in optical laser devices.
7. Conclusions
In view of the current manifold applications of organic π-conjugated systems end-capped with electron donors and acceptors, understanding of their structure–property relationships seems to be crucial. Fundamental aspects of the property tuning in push–pull molecules summarized in this article are primarily aimed at the design of new D-π-A systems with improved/tailored properties such as FMO levels and electronic absorption/emission behaviour. The property tuning has been demonstrated by variations of particular D, π and A components of a push–pull molecule. The currently well-established and easily accessible electron donating groups involve aromatic amines (typically N,N-dimethylanilino (DMA), N,N-diphenylanilino (DPA), julolidine, carbazole), electron rich five-membered heterocycles such as thiophene or pyrrole (auxiliary donors), N,N-dialkylaminothiophene (typically piperidylthiophene (PIT)), tetrathiafulvalene (TTF) and metallocenes (typically ferrocene, dual donor). The main advantage of the amino-derived electron donors can be seen in their highly positive mesomeric effects (Table 1), synthetic availability and modularity and the possibility of a substitution with long alkyl chains, which supports good solubility of the chromophore (the eternal problem of push–pull molecules). Besides these “standard donors”, proaromatic electron donors such as 4-pyran-4-ylidine, pyridine-4-ylidine and dithiafulvene were introduced in the early 90’s. In a combination with strong acceptors, these moieties impart very strong ICT into push–pull molecules. An overview of powerful electron donors is shown in Fig. 43. | ||
| Fig. 43 Representative electron donors in push–pull molecules. | ||
Property tuning can also be achieved by varying the π-conjugated backbone connecting the D and A parts. The π-linker is most commonly constructed by a combination of multiple bonds and (hetero)aromatic units. Whereas the D–A interaction across a polyene is generally high, such D–π–A molecules often suffer from thermal/chemical instability and geometric isomerization. On the other hand, rod-like polyynes are more stable but “less transparent”, due to the presence of more electronegative sp-hybridized carbons. A polarization of aromatic units (typically 1,4-phenylene unit) is accompanied by a loss of aromatic stabilization, which is directed against an efficient ICT. Hence, heteroaromatic/proaromatic units became more and more frequently employed as parts of the π-linker. In this respect, 2,5-thienylene and related spacers played very prominent role and became recognized as easily-accessible, polarizable and thus very popular π-conjugated bridges. In any case the overall performance of the given π-linker must be elucidated in terms of the D/A pair attached, its length, composition, planarity and degree of BLA.
As a matter of fact, electron withdrawing groups in push–pull molecules are more diverse than electron donors. Besides the common cyano and nitro groups, the modern push–pull molecules are equipped with more complex withdrawing moieties (Fig. 44). For instance, replacing the cyano by dicyanovinyl (DCV) acceptor groups may shift the longest-wavelength absorption maxima by more than 100 nm and narrowed the HOMO–LUMO gap by up to 1 eV. Malonic acid-derived acceptors such as DCV, cyanoacetic (cyanoacrylic) acid or (thio)barbituric acids (BA and TBA) proved to be a very powerful family of acceptors used in NLO or OPV devices. 1,3-Dicarbonyl compounds represent another more complex acceptor units. Indane-1,3-dione belongs to this type of acceptor, in which the withdrawing ability can further be tailored by appending one/two additional DCV units or a sulfoxide group. Moreover, both types of these acceptor units can be easily introduced via Knoevenagel condensation with donor-substituted carbonyl compounds.
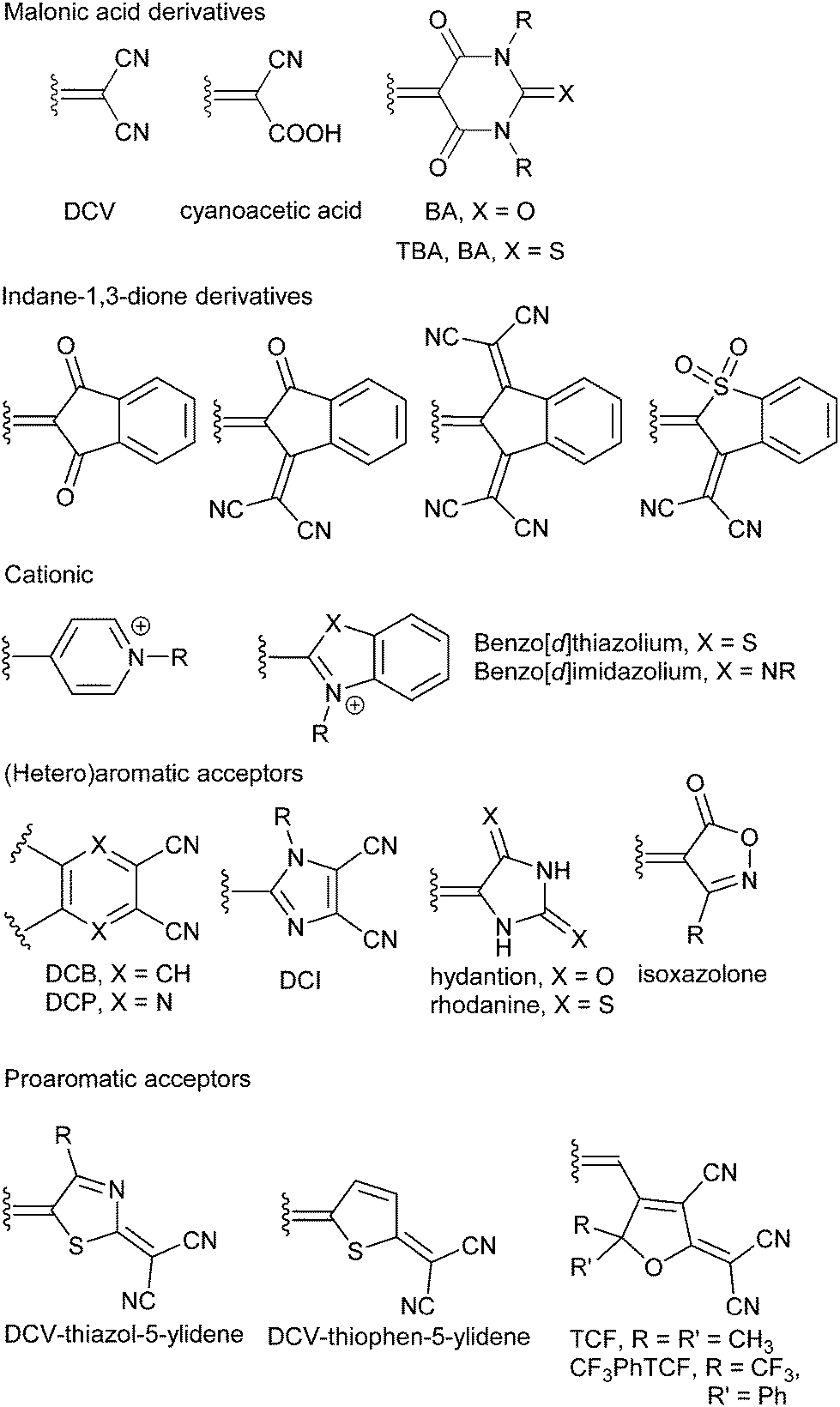 | ||
| Fig. 44 Electron withdrawing moieties applied as acceptors in push–pull molecules. | ||
In principle, almost all (hetero)aromatic scaffolds can be used as more or less efficient electron acceptors if properly substituted and arranged. The most widely employed are electron poor six-membered heterocycles such as (di)azines. Their electron accepting behaviour can be finely tuned by protonation, alkylation or (transition) metal coordination. Pyridinium and quaternized benzo[d]imidazole and benzo[d]thiazole derivatives are well known and easily available. However, the ionic chromophore, with its solubility limited to only polar solvents, may represent an obstacle to its further (opto)electronic applications. We have recently introduced pyrazine/benzene-derived DCP/DCB electron acceptors on which X-shaped push–pull chromophores can be built. Dicyanoimidazole (DCI) can also be used in Y-shaped push–pull molecules, where it is considered as an acceptor with moderate withdrawing character. Five-membered heterocycles such as hydantoin and rhodanine are much powerful electron acceptors than imidazole. The chalcogen effect (O → S replacement) in these units is even more pronounced than in BA → TBA derivatives. The isoxazolone unit also showed a noticeable electron withdrawing ability. The DCV unit connected to a quinoid scaffold (typically benzoquionone) has been demonstrated as a powerful electron acceptor moiety a long time ago. However, appending the DCV unit to a heteroaromatic ring such as thiazol, thiophene or furan afforded a new class of very powerful proaromatic acceptors. These units showed considerably enhanced electron withdrawing ability compared to all preceding acceptors. The fine property tuning of the tricyanofuran (TCF) unit by subtle structural changes further enhances its accepting behaviour as shown by CF3PhTCF.
Acknowledgements
This work was supported by the Czech Science Foundation (13-01061S). This review is dedicated to Prof. François Diederich who has introduced me to the fantastic world of push–pull molecules.Notes and references
- (a) Special issue on “Organic electronics and optoelectronics”, ed. S. R. Forrest and M. E. Thompson, Chem. Rev., 2007, 107, 923 CrossRef CAS; (b) Special issue on “Materials for electronics”, ed. R. D. Miller and E. A. Chandross, Chem. Rev., 2010, 110, 1 CrossRef CAS PubMed; (c) Special issue on “Molecular electronics – from a visionary concept towards reality”, ed. M. Mayor, Chimia, 2010, 64, 348 CAS.
- Special issue on “Molecular conductors”, ed. P. Batail, Chem. Rev., 2004, 104, 4887 CrossRef CAS.
- (a) Special issue on “Organic photovoltaics”, ed. J. L. Bredas and J. R. Durrant, Acc. Chem. Res., 2009, 42, 1689 CrossRef CAS PubMed; (b) Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245 RSC.
- Special issue on “Solar photon conversion”, ed. A. J. Nozik and J. R. Miller, Chem. Rev., 2010, 110, 6443 CrossRef CAS PubMed.
- S. Allard, M. Forster, B. Souharce, H. Thiem and U. Scherf, Angew. Chem., Int. Ed., 2008, 47, 4070 CrossRef CAS PubMed.
- Y. Ohmori, Laser Photonics Rev., 2009, 4, 300 CrossRef.
- A. W. Hains, Z. Liang, M. A. Woodhouse and B. A. Gregg, Chem. Rev., 2010, 110, 6689 CrossRef CAS PubMed.
- (a) Y. Wu and W. Zhu, Chem. Soc. Rev., 2013, 42, 2039 RSC; (b) J. N. Clifford, E. Martínez-Ferrero, A. Viterisi and E. Palomares, Chem. Soc. Rev., 2011, 40, 1635 RSC; (c) C. Duan, K. Zhang, C. Zhong, F. Huang and Y. Cao, Chem. Soc. Rev., 2013, 42, 9071 RSC; (d) M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453 RSC.
- C. Duan, F. Huang and Y. Cao, J. Mater. Chem., 2012, 22, 10416 RSC.
- (a) M. Kivala and F. Diederich, Acc. Chem. Res., 2009, 42, 235 CrossRef CAS PubMed; (b) S. Kato and F. Diederich, Chem. Commun., 2010, 46, 1994 RSC; (c) H. Meier, Angew. Chem., Int. Ed., 2005, 44, 2482 CrossRef CAS PubMed; (d) M. Kivala and F. Diederich, Pure Appl. Chem., 2008, 80, 411 CrossRef CAS.
- Handbook of thiophene-based materials, applications in organic electronics and photonics, ed. I. F. Perepichka and D. F. Perepichka, Wiley, Chicester, 2009, vol. 1 and 2 Search PubMed.
- (a) R. Andreu, L. Carrasquer, S. Franco, J. Garín, J. Orduna, N. M. de Baroja, R. Alicante, B. Villacampa and M. Allain, J. Org. Chem., 2009, 74, 6647 CrossRef CAS PubMed; (b) R. Andreu, E. Galán, J. Garín, V. Herrero, E. Lacarra, J. Orduna, R. Alicante and B. Villacampa, J. Org. Chem., 2010, 75, 1684 CrossRef CAS PubMed; (c) N. Faux, B. Caro, F. Robin-Le Guen, P. Le Poul, K. Nakatani and E. Ishow, J. Organomet. Chem., 2005, 690, 4982 CrossRef CAS PubMed; (d) S. Gauthier, N. Vologdin, S. Achelle, A. Barsella, B. Caro and F. Robin-Le Guen, Tetrahedron, 2013, 69, 8392 CrossRef CAS PubMed.
- For reviews on metallocenes as donors see: (a) N. J. Long, Angew. Chem., Int. Ed. Engl., 1995, 34, 21 CrossRef CAS; (b) H. S. Nalwa, Appl. Organomet. Chem., 1991, 5, 349 CrossRef CAS.
- (a) J. Kulhánek, F. Bureš, W. Kuznik, I. V. Kityk, T. Mikysek and A. Růžička, Chem. - Asian J., 2013, 8, 465 CrossRef PubMed; (b) S. Barlow, H. E. Bunting, C. Ringham, J. C. Green, G. U. Bublitz, S. G. Boxer, J. W. Perry and S. R. Marder, J. Am. Chem. Soc., 1999, 121, 3715 CrossRef CAS; (c) S. Salman, J.-L. Brédas, S. R. Marder, V. Coropceanu and S. Barlow, Organometallics, 2013, 32, 6061 CrossRef CAS; (d) T. L. Kinnibrugh, S. Salman, Y. A. Getmanenko, V. Coropceanu, W. W. Porter III, T. V. Timofeeva, A. J. Matzger, J.-L. Brédas, S. R. Marder and S. Barlow, Organometallics, 2009, 28, 1350 CrossRef CAS PubMed.
- (a) S. Achelle, C. Baudequin and N. Plé, Dyes Pigm., 2013, 98, 575 CrossRef CAS PubMed; (b) S. Achelle, N. Plé and A. Turck, RSC Adv., 2011, 1, 364 RSC; (c) S. Achelle and N. Plé, Curr. Org. Synth., 2012, 9, 163 CrossRef CAS.
- (a) P. Hrobárik, V. Hrobáriková, I. Sigmundová, P. Zahradník, M. Fakis, I. Polyzos and P. Persephonis, J. Org. Chem., 2011, 76, 8726 CrossRef PubMed; (b) V. Hrobáriková, P. Hrobárik, P. Gajdoš, I. Fitilis, M. Fakis, P. Persephonis and P. Zahradník, J. Org. Chem., 2010, 75, 3053 CrossRef PubMed.
- J. Kulhánek and F. Bureš, Beilstein J. Org. Chem., 2012, 8, 25 CrossRef PubMed.
- (a) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS; (b) O. Pytela, Collect. Czech. Chem. Commun., 1996, 61, 704 CrossRef CAS.
- (a) M. A. Ramírez, A. M. Cuadro, J. Alvarez-Builla, O. Castaño, J. L. Andrés, F. Mendicuti, K. Clays, I. Asselberghs and J. J. Vaquero, Org. Biomol. Chem., 2012, 10, 1659 RSC; (b) M. A. Ramirez, T. Cañeque, A. M. Cuadro, F. Mendicuti, K. Clays, I. Asselbergh and J. J. Vaquero, ARKIVOC, 2011, iii, 140 Search PubMed.
- (a) W. Wu, C. Wang, C. Zhong, C. Ye, G. Qui, J. Qin and Z. Li, Polym. Chem., 2013, 4, 378 RSC; (b) J. Zhu, C. Lu, Y. Cui, C. Zhang and G. Lu, J. Chem. Phys., 2010, 133, 244503 CrossRef PubMed.
- Benzene-derived X-shaped chromophores: (a) J. A. Marsden, J. J. Miller, L. D. Shirtcliff and M. M. Haley, J. Am. Chem. Soc., 2005, 127, 2464 CrossRef CAS PubMed; (b) E. L. Spitler, L. D. Shirtcliff and M. M. Haley, J. Org. Chem., 2007, 72, 86 CrossRef CAS PubMed; (c) D. T. Chase, B. S. Young and M. M. Haley, J. Org. Chem., 2011, 76, 4043 CrossRef CAS PubMed; (d) H. Kang, G. Evmenenko, P. Dutta, K. Clays, K. Song and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 6194 CrossRef CAS PubMed.
- Ethene-derived X-shaped chromophores: (a) N. K. Pahadi, D. H. Camacho, I. Nakamura and Y. Yamamoto, J. Org. Chem., 2006, 71, 1152 CrossRef CAS PubMed; (b) N. N. P. Moonen, C. Boudon, J.-P. Gisselbrecht, P. Seiler, M. Gross and F. Diederich, Angew. Chem., Int. Ed., 2002, 41, 3044 CrossRef CAS; (c) N. N. P. Moonen, W. C. Pomerantz, R. Gist, C. Boudon, J.-P. Gisselbrecht, T. Kawai, A. Kishioka, M. Gross, M. Irie and F. Diederich, Chem. - Eur. J., 2005, 11, 3325 CrossRef CAS PubMed; (d) F. Bureš, W. B. Schweizer, J. C. May, C. Boudon, J.-P. Gisselbrecht, M. Gross, I. Biaggio and F. Diederich, Chem. - Eur. J., 2007, 13, 5378 CrossRef PubMed.
- Heteroaromate-derived X-shaped chromophores: (a) F. Bureš, H. Čermáková, J. Kulhánek, M. Ludwig, W. Kuznik, I. V. Kityk, T. Mikysek and A. Růžička, Eur. J. Org. Chem., 2012, 529 CrossRef; (b) Y. Zhao, C. Zhang, K. F. Chin, O. Pytela, G. Wei, H. Liu, F. Bureš and Z. Jiang, RSC Adv., 2014, 4, 30062 RSC.
- (a) Special issue on “Optical nonlinearities in chemistry”, ed. D. Burland, Chem. Rev., 1994, 94, 1 CrossRef; (b) G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245 CrossRef CAS PubMed; (c) Y. Kawabe, H. Ikeda, T. Sakai and K. Kawasaki, J. Mater. Chem., 1992, 2, 1025 RSC; (d) S. R. Marder and J. W. Perry, Adv. Mater., 1993, 5, 804 CrossRef CAS.
- (a) P. A. Sullivan and L. R. Dalton, Acc. Chem. Res., 2010, 43, 10 CrossRef CAS PubMed; (b) M. Stähelin, B. Zysset, M. Ahlheim, S. R. Marder, P. V. Bedworth, C. Runser, M. Barzoukas and A. Fort, J. Opt. Soc. Am. B, 1996, 13, 2401 CrossRef.
- F. Chen, J. Zhang and X. Wan, Chem. - Eur. J., 2012, 18, 4558 CrossRef CAS PubMed.
- (a) B. J. Coe, Chem. - Eur. J., 1999, 5, 2464 CrossRef CAS; (b) I. Asselberghs, K. Clays, A. Persoons, M. D. Ward and J. McCleverty, J. Mater. Chem., 2004, 14, 2831 RSC.
- F. M. Raymo and M. Tomasulo, Chem. - Eur. J., 2006, 12, 3186 CrossRef CAS PubMed.
- (a) C. Reichardt, Solvents and solvent effects in organic chemistry, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) F. Bureš, O. Pytela, M. Kivala and F. Diederich, J. Phys. Org. Chem., 2011, 24, 274 CrossRef; (c) F. Bureš, O. Pytela and F. Diederich, J. Phys. Org. Chem., 2009, 22, 155 CrossRef.
- (a) M. G. Kuzyk, J. Mater. Chem., 2009, 19, 7444 RSC; (b) Y. Sheng, Y. Jiang and X.-C. Wang, J. Chem. Soc., Faraday Trans., 1998, 94, 47 RSC; (c) J. Y. Lee, K. S. Kim and B. J. Mhin, J. Chem. Phys., 2001, 115, 9484 CrossRef CAS PubMed; (d) T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498 CrossRef CAS; (e) A. Datta and S. Pal, THEOCHEM, 2005, 715, 59 CrossRef CAS PubMed; (f) F. Bureš, Chem. Listy, 2013, 107, 834 Search PubMed.
- (a) J. Roncali, Macromol. Rapid Commun., 2007, 28, 1761 CrossRef CAS; (b) J. Roncali, Chem. Rev., 1997, 97, 173 CrossRef CAS PubMed; (c) H. A. M. van Mullekom, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Mater. Sci. Eng., R, 2001, 32, 1 CrossRef; (d) D. F. Perepichka and M. R. Bryce, Angew. Chem., Int. Ed., 2005, 44, 5370 CrossRef CAS PubMed.
- J. Kulhánek, F. Bureš, O. Pytela, T. Mikysek, J. Ludvík and A. Růžička, Dyes Pigm., 2010, 85, 57 CrossRef PubMed.
- J. Kulhánek, F. Bureš and M. Ludwig, Beilstein J. Org. Chem., 2009, 5, 11 Search PubMed.
- A. A. Isse and A. Gennaro, J. Phys. Chem. B, 2010, 114, 7894 CrossRef CAS PubMed.
- J. Kulhánek, F. Bureš, A. Wojciechowski, M. Makowska-Janusik, E. Gondek and I. V. Kityk, J. Phys. Chem. A, 2010, 114, 9440 CrossRef PubMed.
- F. Würthner, F. Effenberger, R. Wortmann and P. Krämer, Chem. Phys., 1993, 173, 305 CrossRef.
- (a) A. E. Stiegman, E. Graham, K. J. Perry, L. R. Khundkar, L.-T. Cheng and J. W. Perry, J. Am. Chem. Soc., 1991, 113, 7658 CrossRef CAS; (b) C. Dehu, F. Meyers and J. L. Brédas, J. Am. Chem. Soc., 1993, 115, 6198 CrossRef CAS.
- S. R. Marder, L.-T. Cheng and B. G. Tiemann, J. Chem. Soc., Chem. Commun., 1992, 672 RSC.
- (a) L.-T. Cheng, W. Tam, S. H. Stevenson, G. R. Meredith, G. Rikken and S. R. Marder, J. Phys. Chem., 1991, 95, 10631 CrossRef CAS; (b) L.-T. Cheng, W. Tam, S. R. Marder, A. E. Stiegman, G. Rikken and C. W. Spangler, J. Phys. Chem., 1991, 95, 10643 CrossRef CAS; (c) S. R. Marder, C. B. Gorman, B. G. Tiemann and L.-T. Cheng, J. Am. Chem. Soc., 1993, 115, 3006 CrossRef CAS; (d) G. Bourhill, J.-L. Brédas, L.-T. Cheng, S. R. Marder, F. Meyers, J. W. Perry and B. G. Tiemann, J. Am. Chem. Soc., 1994, 116, 2619 CrossRef CAS.
- (a) M. Blanchard-Desce, V. Alain, P. V. Bedworth, S. R. Marder, A. Fort, C. Runser, M. Barzoukas, S. Lebus and R. Wortmann, Chem. - Eur. J., 1997, 3, 1091 CrossRef CAS; (b) E. Genin, V. Hugues, G. Clermont, C. Herbivo, M. C. R. Castro, A. Comel, M. M. M. Raposo and M. Blanchard-Desce, Photochem. Photobiol. Sci., 2012, 11, 1756 RSC.
- A. Abbotto, L. Beverina, R. Bozio, S. Bradamante, C. Ferrante, G. A. Pagani and R. Signorini, Adv. Mater., 2000, 12, 1963 CrossRef CAS.
- J. Zhang, L. Yang, M. Zhang and P. Wang, RSC Adv., 2013, 3, 6030 RSC.
- R. Tarsang, V. Promarak, T. Sudyoadsuk, S. Namuangruk and S. Jungsuttiwong, J. Photochem. Photobiol., A, 2014, 273, 8 CrossRef CAS PubMed.
- M.-B. S. Kirketerp, M. Å. Petersen, M. Wanko, H. Zettergren, A. Rubio, M. B. Nielsen and S. B. Nielsen, ChemPhysChem, 2010, 11, 2495 CrossRef CAS PubMed.
- (a) M. Makowska-Janusik, I. V. Kityk, J. Kulhánek and F. Bureš, J. Phys. Chem. A, 2011, 115, 12251 CrossRef CAS PubMed; (b) A. Plaquet, B. Champagne, J. Kulhánek, F. Bureš, E. Bogdan, F. Castet, L. Ducasse and V. Rodriguez, ChemPhysChem, 2011, 12, 3245 CrossRef CAS PubMed; (c) N. Almonasy, F. Bureš, J. Kulhánek and O. Machalický, J. Mater. Sci. Eng. A, 2011, 1, 146 CAS; (d) M. Nepraš, N. Almonasy, F. Bureš, J. Kulhánek, M. Dvořák and M. Michl, Dyes Pigm., 2011, 91, 466 CrossRef PubMed; (e) M. Danko, P. Hrdlovič, J. Kulhánek and F. Bureš, J. Fluoresc., 2011, 21, 1779 CrossRef CAS PubMed; (f) F. Bureš, J. Kulhánek, T. Mikysek, J. Ludvík and J. Lokaj, Tetrahedron Lett., 2010, 51, 2055 CrossRef PubMed.
- (a) S. Yang and M. Kertesz, J. Phys. Chem. A, 2006, 110, 9771 CrossRef CAS PubMed; (b) D. Jacquemin and C. Adamo, J. Chem. Theory Comput., 2011, 7, 369 CrossRef CAS; (c) M. Medveď and D. Jacquenim, Chem. Phys., 2013, 415, 196 CrossRef PubMed; (d) C. Maertens, C. Detrembleur, P. Dubois, R. Jérôme, C. Boutton, A. Persoons, T. Kogej and J. L. Brédas, Chem. - Eur. J., 1999, 5, 369 CrossRef CAS.
- A. M. Moran, M. Blanchard-Desce and A. M. Kelly, Chem. Phys. Lett., 2002, 358, 320 CrossRef CAS.
- J. Garín, J. Orduna, J. I. Rupérez, R. Alcalá, B. Villacampa, C. Sánchez, N. Martín, J. L. Segura and M. Gonzáles, Tetrahedron Lett., 1998, 39, 3577 CrossRef.
- C. R. Moylan, R. J. Twieg, V. Y. Lee, S. A. Swanson, K. M. Betterton and R. D. Miller, J. Am. Chem. Soc., 1993, 115, 12599 CrossRef CAS.
- C. Coluccini, A. K. Sharma, M. Caricato, A. Sironi, E. Cariati, S. Righetto, E. Tordin, C. Botta, A. Forni and D. Pasini, Phys. Chem. Chem. Phys., 2013, 15, 1666 RSC.
- J. C. May, I. Baggio, F. Bureš and F. Diederich, Appl. Phys. Lett., 2007, 90, 251106 CrossRef PubMed.
- E. Zojer, D. Beljonne, P. Pacher and J.-L. Brédas, Chem. - Eur. J., 2004, 10, 2668 CrossRef CAS PubMed.
- (a) I. D. L. Albert, T. J. Marks and M. A. Ratner, J. Am. Chem. Soc., 1997, 119, 6575 CrossRef CAS; (b) S. Bradamante, A. Facchetti and G. A. Pagani, J. Phys. Org. Chem., 1997, 10, 514 CrossRef CAS; (c) J. L. Brédas, J. Chem. Phys., 1985, 82, 3808 CrossRef PubMed.
- J. Kulhánek, F. Bureš, J. Opršal, W. Kuznik, T. Mikysek and A. Růžička, Asian J. Org. Chem., 2013, 2, 422 CrossRef.
- (a) A. K.-Y. Jen, V. P. Rao, K. Y. Wong and K. J. Drost, J. Chem. Soc., Chem. Commun., 1993, 90 RSC; (b) V. P. Rao, Y. M. Cai and A. K.-Y. Jen, J. Chem. Soc., Chem. Commun., 1994, 1689 RSC.
- K. Y. Wong, A. K.-Y. Jen, V. P. Rao and K. J. Drost, J. Chem. Phys., 1994, 100, 6818 CrossRef CAS PubMed.
- C. R. Moylan, B. J. McNelis, L. C. Nathan, M. A. Marques, E. L. Hermstadt and B. A. Brichler, J. Org. Chem., 2004, 69, 8239 CrossRef CAS PubMed.
- (a) M. M. Oliva, J. Casado, M. M. M. Raposo, A. M. C. Fonseca, H. Hartmann, V. Hernández and J. T. L. Navarrete, J. Org. Chem., 2006, 71, 7509 CrossRef PubMed; (b) M. M. M. Raposo and G. Kirsch, Tetrahedron, 2003, 59, 4891 CrossRef CAS; (c) M. M. M. Raposo, A. M. C. Fonseca and G. Kirsch, Tetrahedron, 2004, 60, 4071 CrossRef CAS PubMed; (d) A. Wojciechowski, M. M. M. Raposo, M. C. R. Castro, W. Kuznik, I. Fuks-Janczarek, M. Pokladko-Kowar and F. Bureš, J. Mater. Sci.: Mater. Electron., 2014, 25, 1745 CrossRef CAS PubMed.
- P. V. Bedworth, Y. Cai, A. Jen and S. R. Marder, J. Org. Chem., 1996, 61, 2242 CrossRef CAS.
- I. D. L. Albert, T. J. Marsk and M. A. Ratner, J. Am. Chem. Soc., 1997, 119, 6575 CrossRef CAS.
- P. R. Varanasi, A. K.-Y. Jen, J. Chandrasekhar, I. N. N. Namboothiri and A. Rathna, J. Am. Chem. Soc., 1996, 118, 12443 CrossRef.
- B. O’Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- D. Casanova, ChemPhysChem, 2011, 12, 2979 CrossRef CAS PubMed.
- D. P. Hagberg, T. Marinado, K. M. Karlsson, K. Nonomura, P. Qin, G. Boschloo, T. Brinck, A. Hagfeldt and L. Sun, J. Org. Chem., 2007, 72, 9550 CrossRef CAS PubMed.
- (a) S. H. Kim, H. W. Kim, C. Sakong, J. Namgoong, S. W. Park, M. J. Ko, C. H. Lee, W. I. Lee and J. P. Kim, Org. Lett., 2011, 13, 5784 CrossRef CAS PubMed; (b) H. Tian, X. Yang, R. Chen, Y. Pan, L. Li, A. Hagfeldt and L. Sun, Chem. Commun., 2007, 3741 RSC.
- R. Li, X. Lv, D. Shi, D. Zhou, Y. Cheng, G. Zhang and P. Wang, J. Phys. Chem. C, 2009, 113, 7469 CAS.
- S. R. Marder, D. N. Beratan and L.-T. Cheng, Science, 1991, 252, 103 CAS.
- Characterization techniques and tabulations for organic nonlinear optical materials, ed. M. G. Kuzyk and C. W. Dirk, Marcel Dekker, New York, 1998 Search PubMed.
- (a) Y.-L. Wu, F. Bureš, P. D. Jarowski, W. B. Schweizer, C. Boudon, J.-P. Gisselbrecht and F. Diederich, Chem. - Eur. J., 2010, 16, 9592 CrossRef CAS PubMed; (b) F. Bureš, W. B. Schweizer, C. Boudon, J.-P. Gisselbrecht, M. Gross and F. Diederich, Eur. J. Org. Chem., 2008, 994 CrossRef.
- T. Kawase, M. Wakabayashi, C. Takahashi and M. Oda, Chem. Lett., 1997, 1055 CrossRef CAS.
- A. J. Kay, A. D. Woolhouse, Y. Zhao and K. Clays, J. Mater. Chem., 2004, 14, 1321 RSC.
- A. Abbotto, L. Beverina, S. Bradamante, A. Fachetti, C. Klein, G. A. Pagani, M. Redi-Abshiro and R. Wortmann, Chem. - Eur. J., 2003, 9, 1991 CrossRef CAS PubMed.
- S. Alías, R. Andreu, M. J. Blesa, S. Franco, J. Garín, A. Gragera, J. Orduna, P. Romero, B. Villacampa and M. Allain, J. Org. Chem., 2007, 72, 6440 CrossRef PubMed.
- R. Andreu, M. A. Cerdán, S. Franco, J. Garín, A. B. Marco, J. Orduna, D. Palomas, B. Villacampa, R. Alicante and M. Allain, Org. Lett., 2008, 10, 4963 CrossRef CAS PubMed.
- R. Andreu, E. Galán, J. Orduna, B. Villacampa, R. Alicante, J. T. L. Navarrete, J. Casado and J. Garín, Chem. - Eur. J., 2011, 17, 826 CrossRef CAS PubMed.
- (a) R. Gompper, E. Kutter and H.-U. Wagner, Angew. Chem., Int. Ed. Engl., 1966, 5, 517 CrossRef CAS; (b) R. Gompper, H.-U. Wagner and E. Kutter, Chem. Ber., 1968, 101, 4123 CrossRef CAS.
- S. Inoue, Y. Aso and T. Otsubo, Chem. Commun., 1997, 1105 RSC.
- S. Inoue, S. Mikami, Y. Aso, T. Otsubo, T. Wada and H. Sasabe, Synth. Met., 1997, 84, 395 CrossRef CAS.
- A. S. Batsanov, M. R. Bryce, M. A. Coffin, A. Green, R. E. Hester, J. A. K. Howard, I. K. Lednev, N. Martín, A. J. Moore, J. N. Moore, E. Ortí, L. Sánchez, M. Savirón, P. M. Viruela, R. Viruela and T.-Q. Ye, Chem. - Eur. J., 1998, 4, 2580 CrossRef CAS.
- R. Andreu, J. Garín, J. Orduna, R. Alcalá and B. Villacampa, Org. Lett., 2003, 5, 3143 CrossRef CAS PubMed.
- S. Alías, R. Andreu, M. J. Blesa, M. A. Cerdán, S. Franco, J. Garín, C. López, J. Orduna, J. Sanz, R. Alicante, B. Villacampa and M. Allain, J. Org. Chem., 2008, 73, 5890 CrossRef PubMed.
- R. Andreu, M. J. Blesa, L. Carrasquer, J. Garín, J. Orduna, B. Villacampa, R. Alcalá, J. Casado, M. C. R. Delgado, J. T. L. Navarrete and M. Allain, J. Am. Chem. Soc., 2005, 127, 8835 CrossRef CAS PubMed.
- M. G. Vivas, D. L. Silva, J. Malinge, M. Boujtita, R. Zaleśny, W. Bartkowiak, H. Ågren, S. Canuto, L. De Boni, E. Ishow and C. R. Mendonca, Sci. Rep., 2014, 4, 4447 Search PubMed.
- C. Herbivo, A. Comel, G. Kirsch, A. M.C. Fonseca, M. Belsley and M. M. M. Raposo, Dyes Pigm., 2010, 86, 217 CrossRef CAS PubMed.
- J.-M. Raimundo, P. Blanchard, I. Ledoux-Rak, R. Hierle, L. Michaux and J. Roncali, Chem. Commun., 2000, 1597 RSC.
- L. Dokládalová, F. Bureš, W. Kuznik, I. V. Kityk, A. Wojciechowski, T. Mikysek, N. Almonasy, M. Ramaiyan, Z. Padělková, J. Kulhánek and M. Ludwig, Org. Biomol. Chem., 2014, 12, 5517 Search PubMed.
- M. Klikar, F. Bureš, O. Pytela, T. Mikysek, Z. Padělková, A. Barsella, K. Dorkenoo and S. Achelle, New J. Chem., 2013, 37, 4230 RSC.
- S. Achelle, S. Kahlal, A. Barsella, J.-Y. Saillard, X. Che, J. Vallet, F. Bureš, B. Caro and F. Robin-le Guen, Dyes Pigm., 2015, 113, 562 CrossRef CAS PubMed.
- B. J. Coe, J. A. Harris, J. J. Hall, B. S. Brunschwig, S.-T. Hung, W. Libaers, K. Clays, S. J. Coles, P. N. Horton, M. E. Light, M. B. Hursthouse, J. Garín and J. Orduna, Chem. Mater., 2006, 18, 5907 CrossRef CAS.
- F. Quist, C. M. L. Vande Velde, D. Didier, A. Teshome, I. Asselberghs, K. Clays and S. Sergeyev, Dyes Pigm., 2009, 81, 203 CrossRef CAS PubMed.
- H. Ikeda, Y. Kawabe, T. Sakai and K. Kawasaki, Chem. Lett., 1989, 1803 CrossRef CAS.
- Y. Liao, B. E. Eichinger, K. A. Firestone, M. Haller, J. Luo, W. Kaminsky, J. B. Benedict, P. J. Reid, A. K.-Y. Jen, L. R. Dalton and B. H. Robinson, J. Am. Chem. Soc., 2005, 127, 2758 CrossRef CAS PubMed.
- F. Würthner, C. Thalacker, R. Matschiner, K. Lukaszuk and R. Wortmann, Chem. Commun., 1998, 1739 RSC.
- H. Bürckstümmer, E. V. Tulyakova, M. Deppisch, M. R. Lenze, N. M. Kronenberg, M. Gsänger, M. Stolte, K. Meerholz and F. Würthner, Angew. Chem., Int. Ed., 2011, 50, 11628 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11264d |
| This journal is © The Royal Society of Chemistry 2014 |