
Reinforcing efficiency and compatibilizing effect of sol–gel derived in situ silica for natural rubber/chloroprene rubber blends†
Abstract
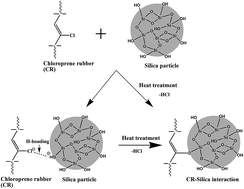
Nano silica is grown, in situ, in natural rubber (NR)/chloroprene rubber (CR) blends, by the soaking sol–gel method. Much better silica dispersion in the rubber blends is achieved following this technique in comparison to the rubber blends with externally filled silica at the same filler loading and same blend composition. This leads to a significant improvement in modulus, tensile strength and dynamic mechanical properties of all the in situ silica filled composites relative to externally filled composites. Additionally, analysis of glass transition temperature (Tg) values reveals that compatibility of NR and CR in the blend is enhanced when silica is incorporated in situ which in turn contributes to improving the physical properties of the composites. This enhancement in the compatibility of rubber blends is attributed to the preferential accumulation of in situ silica at the interphase of the two constituent rubbers. The best mechanical properties are shown by the in situ filled composite with NR/CR at a 40/60 blend ratio. This result is in agreement with the rheological properties, thermal properties and viscoelastic behaviors of this particular composite. The ultimate properties of the composites are found to be governed by the blend composition, blend compatibility and state of filler dispersion, in addition to the filler content.
 Please wait while we load your content...
Please wait while we load your content...

