
Fluorescent micellar nanoparticles by self-assembly of amphiphilic, nonionic and water self-dispersible polythiophenes with “hairy rod” architecture†
Luminita Cianga‡
a,
Anca-Dana Bendrea‡a,
Nicusor Fiferea,
Loredana Elena Nitaa,
Florica Dorofteia,
Didem Agb,
Muharrem Selecib,
Suna Timur*b and
Ioan Cianga*a
a“Petru Poni” Institute of Macromolecular Chemistry, 41A Grigore-Ghica Voda Alley, Iasi, 700487, Romania. E-mail: ioanc@icmpp.ro; Fax: +40-232-211299; Tel: +40-232-217454
bDepartment of Biochemistry, Faculty of Science, Ege University, Izmir 35100, Turkey. E-mail: suna.timur@ege.edu.tr; Fax: +90-232-3438624; Tel: +90-232-3115425
First published on 21st October 2014
Abstract
Polymers with “hairy-rod” architecture having oligo/polythiophene (PTh) as main chain and poly (ethylene glycol) (PEG) (Mn = 2000) as flexible side chains were obtained by combining the “macromonomer technique” with specific methods for the synthesis of conjugated polymers. Fluorescent nanoparticles of core–shell type with high colloidal stability were obtained from these water self-dispersible materials by a direct dissolution method in aqueous media. It has been shown that the size and photophysical properties of the micellar nanoparticles in aqueous dispersions as well as the bulk properties of the investigated materials can be tuned by varying the PEG side chain density and by the modality of PEG connection to the PTh main chains. The presence of the PEG shells in the structure of these fluorescent nanoparticles cans suppress the non-specific interactions with biomolecules on the one side and on the other side they work as a biomimetic interface that could facilitate their potential use as cell-imaging agents. The present attempt offers an ease of access alternative to conducting polymer nanoparticle encapsulation in a biocompatible matrix by nanoprecipitation.
Introduction
Recently, there has been considerable effort to synthesize colloidal nanoparticles and to exploit them for various biological applications. Polymer nanoparticles, including polymeric micelles with core–shell structure, self-assembled from amphiphilic copolymers, have demonstrated a very important role in different fields, even in nanomedicine.1 Fluorescent nanoparticles have been particularly useful as probes and photon sources for numerous biological applications2 and polymeric micelles with fluorescent characteristics have attracted tremendous interest in the application of optical imaging for monitoring biological processes.3 As it was recently reported,4 research on semiconducting polymers for biomedical applications expanded greatly in the 1980s since it was found that these materials are compatible with many biological molecules, and impetuous research has been developed in the last decade toward their biomedical uses5 like tissue engineering6 or as nanomedicines.7 Conjugated polymers as fluorescent nanoparticles (CPN) are relatively little addressed,8 even if they can be an appropriate choice for both optoelectronic9 and biomedical applications.10CPN have recently emerged as an alternative promising class of fluorescent probes in cellular imaging. They exhibit remarkable photophysical properties which are the essential requests for cell imaging (stable and bright emission, high quantum yield and large absorption coefficient, high photostability).10 More importantly, these CPN showed any cytotoxicity against different cell lines, suggesting that conjugated polymers are relatively safe for the use in biological applications.6 However, due to their inherent hydrophobicity, the demonstration of CPN as fluorescent probes is still rather limited.8a The water ubiquity in biological context requires finding a way for their transfer in aqueous medium. On the other hand another challenge related to CPN is referring to their obtainment. In principle, such nanoparticles result either directly by polymerization in heterophase systems8b or are produced by post-polymerization methods.8,11 The latter approach has been employed mostly in the form of so-called “nanoprecipitation”11b for which the colloidal stabilization mechanism in the surfactant free dispersions often remained unclear. It was emphasized that without steric or electrostatic stabilization, even for such very diluted dispersions, coagulation should occur on the time scale of seconds.12 In order to mitigate the aspects emphasized above, different strategy related to the conjugated copolymers structure, architecture and preparative procedure must be approached.
As such, conjugated polymers organizing capability through self-assembling in solution13 can be usefully directed by their covalent linking with hydrophilic coil-like polymers in a rod–coil combination with various architectures. The resulting amphiphilic π-conjugated rod–coil molecules could be of great interest as nanofunctional materials, being excellent candidates in creating well-defined, fluorescent micellar structures by self-assembling in aqueous solutions with advantages in terms of environmental friendliness and direct applicability as biomaterials when hydrophilic coil segments like PEG, PNIPAM, etc. are used.8c,14,15
The self-assembly of rod–coil copolymers is fundamentally different from those of conventional coil–coil block-copolymers, the π–π interactions between the conjugated rods providing an additional control factor and functionality.16
Several advantages of the amphiphilic π-conjugated rod–coils are worthy to notice: (i) the capability to form well-ordered structures in selective solvents even at low molecular weights;15 (ii) the increased stiffness of the water insoluble rod results in a decreased critical micellar concentration;17 (iii) supramolecular shape of the formed aggregates in water as well as their stability can be manipulated by the variation of macromolecular architecture.18
For clinical applications, the stability of micelles is one of the major concerns, since they tend to disassemble if the polymer concentration is below the critical micellar concentration. It was shown that for amphiphilic copolymers in selective solvents, near to hydrophilic/lipophilic balance, (HLB), and to staking interactions in the core, the architecture and topology are also very important factors in improving micellar nanoparticles stability.19 In this context, the amphiphilic “molecular brushes”20 become a very attractive option,21 their backbones acting as cross-links on the hydrophobic core surface of the micellar nanoparticles.22 Moreover, in comparison to their linear counterparts, they display extended in vivo circulation times21a and also a much lower critical micellar concentration.1a
On the other hand macromolecules called as “hairy-rods” (see a pictorial drawing in ESI, Fig. S1†) have proved to be particularly successful designs revealing novel types of supramolecular architectures, best described as “molecularly reinforced liquids”.23 “Hairy-rods” concept based on the introduction of conformationally mobile, relatively long, flexible and densely grafted side chains to a rigid π-conjugated backbone has been developed by G. Wegner, which stated that “shape persistence” principle found in the world of biomacromolecules can be also achieved in the conjugated polymers field by direct synthesis.24 Novel polymerisation strategies like controlled free radical polymerizations, allowed the synthesis of various “hairy-rods”,25 as well as of new “molecular brushes”,20 which formerly were obtained only by “macromonomer technique”.26 Recently it was emphasized that “hairy-rods” represent a particular case of “molecular brushes” for which the main chain is already rigid.20b In one of the authors' related reports different monografted or heterografted types of “hairy-rods” were obtained as a result of the combination between hydrophobic conjugated, rigid backbones with hydrophobic/hydrophilic flexible coils.27 As grafted flexible coils, usual polymers like polystyrene,27a,b polytetrahydrofuran,27b polycaprolactone,27c,d or PEG27e,f have been used. The physico-chemical properties of these π-conjugated rod–coil copolymers can be tuned in a versatile manner both in bulk and in solutions.
In general, when stimuli-sensitive polymers were used as the grafted side chains,28 amphiphilic π-conjugated “hairy-rods” with pH,28a,b or thermo-responsiveness were reported.28b–d
With the aim of creating new micelle-forming amphiphilic structures with “hairy rod” topology, with fluorescent characteristics appropriate for bioanalysis and imaging and with enhanced thermodynamic and kinetic stability, we developed our design strategy starting from the point that the thiophene-based materials constitute a rich and homogeneous family of π-conjugated compounds mostly used as active components in organic electronics.29 Also in only few years, thiophene-based materials, which are semiconductors and fluorescent, have become an interdisciplinary field of intensive research targeted to their biomedical applications. Fluorescent oligo- and polythiophenes have been employed as useful probes for imaging ex vivo and in vivo of protein-disordered pathogenic states such as Alzheimer's disease and Parkinson's disease,30 for in vitro non-specific cellular imaging31 and about their anticancer activity has also been reported.31,32 Light-activated antibacterial and antifungal activity33 of some thiophene-based materials as well as active reagents in cancer photodynamic therapy were reported,34 too. Meanwhile electroactive polythiophenes as homopolymers and copolymers or as blends with biocompatible/biodegradable counterparts have been revealed as good candidates for tissue engineering applications.35
Inspired by these considerations and following our previous research interest in the thiophene-containing polymers with designed architectures and tuned photophysical properties,36 as well as in PEG-containing macromonomers,27e,f,37 it seemed appropriate to benefit from the combination through covalent linkage of rigid rod, fluorescent polythiophene with PEG as flexible side chains in a branched topology, having as result polymeric materials able for self-assembling as robust core–shell fluorescent micellar nanoparticles in water and other aqueous media.
Thus, the structure reported in this paper offer a simple and easily accessible alternative to conducting polymer nanoparticles encapsulation in biocompatible matrix by nanoprecipitation.38
As presented in Scheme 1, we engineered “hairy rod” structures having different lengths of the oligo/polythiophene main chains and also different PEG grafting density at a constant PEG length, in order to investigate how their HLB could influence size, shape, micellar stability and photophysical properties of the formed micellar fluorescent nanoparticles obtained by direct dissolution method.39
 | ||
| Scheme 1 Synthesis of thiophene macromonomers and polymers. | ||
To the best of our knowledge, reports on fluorescent micelles obtained by direct water dissolution from “hairy rods” are quite limited,28,40 and only few of them were investigated as fluorescent probes for cell imaging.40a,c
On the other side, unsubstituted polythiophene fluorescent nanoparticles (PThN) dispersed in aqueous media were reported in several papers as being obtained only in the presence of ionic or non-ionic surfactants.41 Other idea for transferring of this fluorescent insoluble polymer in aqueous system in nanoparticles form, which was tackled in the present report, is to use thiophene moiety as a building block for new macromolecular fluorescent surfactant structures.
The well-established concept of aggregation of amphiphilic block copolymers in aqueous solutions was translated to a large and more complex dimension by the amphiphilic “molecular brushes”. They have been recently introduced as a new class of polymeric surfactants.42 Therefore in this study we describe the synthesis and structural characterization of two 2,5-dibrominated thiophene-containing macromonomers of PEG, their derived polymers obtained by Yamamoto dehalogenative polycondensation, as well as a quinquethiophene oligomer and polymer derived therefrom using oxidative polycondensation method (Scheme 1). Thermal behaviour of the synthesized compounds was followed by thermogravimetrical analysis (TGA), differential scanning calorimetry (DSC) or polarising light microscopy (POM) investigations. After demonstration of the micellar nanoparticles formation by spontaneous self-assembling during the direct dissolution in aqueous media as selective solvents for PEG moieties, their size in the formed dispersions was assessed by dynamic light scattering (DLS) and in dry state by atomic force microscopy (AFM). Optical properties in different aqueous systems were evaluated in details by UV-Vis and fluorescence measurements and discussed in relation with the chemical structure and the architecture of the polymers. The high photostability of the formed nanoparticles in water solutions under long time irradiation was also demonstrated and the effect of the nonspecific interactions of the micellar nanoparticles with bovine serum albumin (BSA) on their fluorescence properties was studied as well. Finally, preliminary results on cytotoxicity and on the possibility to use these fluorescent, non-ionic, water self-dispersible thiophenic materials for bioimaging are also presented.
Experimental section
Materials
Poly(ethylene glycol) methyl ether Mn = 2000 (PEG2000) (Aldrich), 3-thiophene carboxylic acid (Aldrich), 3-thiophene acetic acid (Aldrich), N,N′-dicyclohexylcarbodiimide (DCCI) (Merk), 4-dimethylamino pyridine (DMAP) (Aldrich), NiCl2 and Zn (Merck), 2,2-bipyridine (bpy), tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) (Aldrich), 2,2′-bithiophene-5-boronic acid pinacol ester (Aldrich), triphenylphosphine (PPh3) (Fluka), Br2(Fluka), N-bromosuccinimide (NBS) (Aldrich), anhydrous FeCl3 (Aldrich) and hydrazine monohydrate (Aldrich) were used as received. Phosphate buffer saline pH 7.4 (PBS), bovine serum albumin (BSA) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazoliumbromide (MTT) were purchased from Sigma-Aldrich, while sodium dodecyl sulfate (SDS) from Applichem. Cell culture supplies including Dulbecco's Modified Eagle Medium (DMEM from Sigma Aldrich), fetal calf serum (FCS) (PAA Laboratories GmbH), Newborn Calf Serum (NCS) (PAA Laboratories GmbH), and penicillin/streptomycin (P/S) were purchased from (PAA Laboratories GmbH). All the used solvents were purified and dried by usual methods. Water used for the experiments was purified by a Millipore Q plus water purification system (resistance 18.2 MU cm).A-549 (lung cancer) and NIH-3T3 (mouse fibroblast) cell lines were provided from German Collection of Microorganisms and Cell Cultures (DSMZ).
Measurements
1H-NMR spectra were recorded at room temperature on a Bruker Avance DRX-400 spectrometer (400 MHz) as solutions in acetone-d6, CDCl3 and D2O and chemical shifts are reported in ppm and referenced to TMS as internal standard. The relative molecular weights and molecular weight distribution (PDI) were determined by gel permeation chromatography (GPC) using a Waters 515 instrument, THF as the eluent at a flow rate of 0.3 mL min−1 and monodisperse polystyrene standards for the calibration plot.The FTIR spectra were recorded on a Bruker Vertex 70 FTIR spectrometer equipped with a diamond ATR device (Golden Gate, Bruker) in transmission mode, by using KBr pellets.
Measurements of UV-Vis and fluorescence for solutions of the synthesized compounds prepared in Milli-Q water or PBS (25 mM; pH = 7.4), were carried out by using a Specord 200 Analytik Jena spectrophotometer and Perkin Elmer LS 55 apparatus, respectively. Photoirradiation experiments were performed in aqueous solutions of polymers using a 500 W high-pressure mercury arc lamp, and suitable glass filters for the obtainment of the irradiation light. The changes in fluorescence emission intensity were measured for each compound at its main emission maximum over a total 120 min period.
Absolute fluorescence quantum yield (ΦF) of the samples was measured on a FluoroMax-4 spectrofluorometer equipped with a Quanta-phi integrating sphere accessory Horiba Jobin Yvon, by exciting the corresponding compound solutions at its maximum absorption, at room temperature. The solution concentration was optimized to obtain an absorbance around 0.1. The slit widths and detector parameters were optimized for maximize but not saturate the excitation Rayleigh peak, in order to obtain an optimal luminescence signal-to-noise ratio.
DSC experiments were conducted on a Maia DSC 200 F3 apparatus (Netzsch, Germany). Around 10 mg of each sample was weighed in pressed and punched in aluminum crucibles. Nitrogen was used as inert atmosphere at a flow rate of 50 mL min−1. Heating and cooling rates of 3 °C min−1, 5 °C min−1 or 10 °C min−1 were applied.
TGA were performed on STA 449 F1 Jupiter equipment (Netzsch, Germany). Around 10 mg of each sample was weighed in alumina crucibles with no lids. A heating rate of 10 °C min−1 was applied. Nitrogen was used as inert atmosphere at a flow rate of 50 mL min−1.
The melting and cooling of polymer P1 as powder was investigated by POM, using an Olympus BH-2 microscope equipped with a Linkam THMS 600/HSF9I heating stage and TMS91 control unit. The magnification of the pictures was the result of 10× of ocular lenses, 20× of the objective lenses and 3× zoom of the photo camera.
The wide-angle X-ray diffraction (WAXD) experiments were performed on a D8 Advance Bruker AXS diffractometer using a CuKα source with an emission current of 36 mA and a voltage of 30 kV. Scans were collected over the 2θ = 2–40° range.
Particles size measurements were carried out by dynamic light scattering (DLS) using a Malvern Zetasizer Nano ZS instrument equipped with a 4.0 mW He–Ne laser operating at 633 nm and a detection angle of 173°. The intensity weighted mean hydrodynamic size (Z average) and the polydispersity factor were obtained from analysis of the autocorrelation function. Samples were used as prepared, at a concentration of 5 × 10−4 μM based on the repeating unit (RU) without filtration. Only the solvents used for their preparation were filtered through NY-0.45 μm filter to ensure the minimization of dust and other particulates. The reported values represent the average of three measurements made for each sample, at 25 °C with an equilibration time of 5 minutes before starting each measurement.
Synthesis of intermediates and macromonomers
The experimental details for the synthesis of 2,5-dibromothiophene-3-carboxylic acid (1), of macromonomers M1 (ref. 40d and 43) and 5T (ref. 35d and 43b) were previously reported, whereas those related to synthesis of 2,5-dibromothiophene-3-acetic acid (2) and of macromonomer M2, together with 1H-NMR and IR spectra of all of them are given in Supplementary information (ESI†).Synthesis of PEG substituted polythiophenes through Yamamoto coupling (P1 and P2)
A 20 mL three-necked round-bottomed flask equipped with condenser, rubber septum, nitrogen inlet–outlet and magnetic stirrer was charged under nitrogen with 0.31 mmol of macromonomer M1 or M2, 0.002 g (0.015 mmol) NiCl2, 0.0025 g (0.016 mmol) bpy, 0.0083g (0.031 mmol) PPh3, 0.063 g (0.963 mmol) Zn and 1 mL of dry dimethylacetamide. The reaction was performed at 85 °C, under nitrogen. The mixture was stirred at the above-mentioned temperature for 5 days. After that time the polymer was precipitated in a large amount of cold diethyl ether, filtered and dried. The polymer was then redissolved in THF, passed through a silicagel column and reprecipitated in cold diethyl ether.1H-NMR signals, CDCl3, (δ, ppm): P1: 8.27–7.35 ppm (TiH), 4.46–4.21 ppm; (CO–OCH2), 4.21–3.45 ppm (CH2O from PEG), 3.45–3.35 (OCH3); Mn, (GPC) = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700, PDI = 4.18.
700, PDI = 4.18.
P2: 7.7–7 ppm TiH, 4.35–4.17 ppm (CO–OCH2), 4.17–3.42 ppm (CH2O from PEG and Ti–CH2), 3.42–3.2 ppm (OCH3); Mn, (GPC) = 5620, PDI = 1.13.
Synthesis of PEG substituted polythiophene from 5T by chemical oxidative polymerization (P5T)
A 25 mL round bottom flask equipped with magnetic stirrer and a lateral neck with a dropping funnel was used. The system was vacuumed and back-filled with dry nitrogen several times. 1.5 mL CHCl3 and 0.5 g (0.22 mmol) 5T were introduced under inert atmosphere. The mixture was cooled down at 0 °C.0.143 g (0.88 mmol) of anhydrous FeCl3 in 2.5 mL CHCl3 was added dropwise during 20 min. The reaction mixture was stirred at room temperature for 4 days. After that time the polymer was separated by precipitation in diethylether filtered and dried. Further purification was achieved by silicagel column chromatography using CH2Cl2 as eluent. The solution was concentrated at rotaevaporatory and reprecipitated in diethyl ether. Then P5T in solution of CH2Cl2 was treated with hydrazine hydrate for 30 minutes and again reprecipitated in diethyl ether. Thus an undoped polymer was obtained. Mn, (GPC) = 3060, PDI = 1.09.
Preparation of micellar nanoparticles by self-assembly in solutions
Micellar nanoparticles of oligo/polymers were obtained in aqueous solution or PBS (25 mM; pH = 7.4), by self-assembly via the direct dissolution method.14,39 If the calculated values of HLB reported at the repeating units are taken into account (ESI, see Table S1†), this self-assembling method seems to be appropriate for the investigated compounds. A weighted amount of polymers was dissolved in 10 mL of water or PBS (25 mM; pH = 7.4), which self-assembled into micelles with a final concentration of 5 × 10−4 μM based on the repeating unit (RU). The solutions were left overnight for equilibration and the micelles diameter was measured by DLS technique. The micelle solutions were used in DLS without microfiltration.Films preparation and characterization
Drop-casting method was used to obtain thin films of polymers from MilliQ water dilute solutions (5 × 10−4 μM based on RU) on freshly cleaved muscovite mica as substrate. Water was evaporated by freeze-drying method using ALPHA 1-2/LD freeze dryer.Microscopic investigations of the obtained films were performed on an Environmental Scanning Electron Microscope (ESEM) type Quanta 200 operating at 30 kV with secondary and back scattering electrons in high vacuum mode.
AFM micrographs were taken in air, on a SPM SOLVER Pro-M instrument. A NSG10/Au Silicon tip with a 35 nm radius of curvature and 255 kHz oscillation mean frequency was used. The apparatus was operated in semi-contact mode, over a 10 × 10 μm2 scan area, 256 × 256 scan point size images being thus obtained.
Cell cultures
A-549 cells were incubated in DMEM containing 10% FCS and 1.0% P/S. NIH-3T3 cells were cultivated in DMEM containing 10% NCS and 1.0% P/S. All cells were cultivated in medium and incubated with polymers and reagents at 37 °C in humidified environment of 5.0% CO2.Cytotoxicity test
MTT assays were used to determine cytotoxicity of 5T, P5T and P1. Cells were seeded out in 96-well-tissue plates (Sarstedt, USA) in a volume of 100 μL. After 3 days cultivation, the wells were populated confluently. A-549 and NIH-3T3 cells were treated with 5T, P5T and P1 probes at 0.01, 0.05, 0.1, 0.2, 0.3, 0.5, 0.75, 1.0 mg mL−1 concentrations for 2 h. Then the samples were removed completely and cells were incubated with 110 μL per well MTT solution (10%, 5.0 mg mL−1 PBS) in medium for 4 h. While this incubation time the formazan complex was produced inside the cells. To release the purple colored salt 100 μL SDS solution (1.0 g SDS in 10 mL 0.01 M HCl) were added per well and after 24 h of incubation, UV-Vis absorption was measured at 570 nm with 630 nm as reference wavelength using a microplate reader Model 680 (BioRad).Cell imaging
A-549 and NIH-3T3 cells were cultivated 2 days in 96-well plates as described before. After cultivation, 5T, P5T and P1 probes were diluted 1:1 with medium and added into the wells. The cells were incubated 2 h at 37 °C with probes. After that, the cells were washed with PBS for two times. Then, labeled cells were imaged using Olympus IX50 fluorescence microscope equipped with a CCD camera (Olympus C4040).
Results and disscusion
Synthesis, structural characterization and properties in bulk
In previous reports different thiophene-containing macromonomers having polymeric chains attached at the 3-position of the heterocycle35b,44 were synthesized using appropriate thiophene derivatives as initiators for polymerisation of conventional monomers (styrene, ε-caprolactone, methylmethacrylate) by various methods (NMP, CROP ATRP).44a–d,f Other thiophene-functionalised macromonomers were obtained by usual polymers chain-ends functionalization with thiophene.35b,44e In the present work, based on the second method, PEG-based macromonomers functionalized with 2,5-dibromothiophene moieties were firstly prepared. In this aim 2,5-dibromothiophene-3-carboxylic acid (1) and 2,5-dibromothiophene-3 acetic acid (2) were synthesized by bromination of 3-thiophene carboxylic acid and 3-thiophene acetic acid respectively, (Scheme 1). In order to follow the influence of the CH2 moiety presence – between the thiophene ring and the ester function at the third position of the heterocycle – on the photophysical properties of derived polymers, these two thiophene acids were deliberately chosen. The molecular weight of PEG used as side chains was chosen as 2000 Da, so that the theoretically calculated values of HLB for the repeating structural units (RU) of the designed materials (ESI, Table S1†), to be around of 0.9. It was reported that such materials are expected to show fully apparent water solubility.45 On the other hand, it has been emphasized in the literature that water-soluble polymers with molecular weights less than 2500 Da can be more easily cleared from the body through renal way.46Macromonomers M1 and M2 were obtained in the next step by the condensation reaction between commercially available PEG2000 with 1 or 2 through a Steglisch esterification reaction.43
1H-NMR spectra of M1 and M2 in acetone-d6 (ESI, Fig. S2†) confirm the proposed structures.
Because the macromonomers M1 and M2 contain in the 3-position of the thienyl ring an ester group, their further polymerization was performed through a dehalogenative polycondensation developed and reported by Yamamoto.47 This is one of the well-suited methods for synthesis of conjugated polymers from dihaloaromatics that contain an ester group as substitute,48 a functionality that seems to be sensitive to the presence of Grignard reagents.48a
The 1H-NMR spectrum of P1 in CDCl3 (Fig. 1a) shows several groups of peaks in the aromatic region (8.26–7.37 ppm).
 | ||
| Fig. 1 1H-NMR spectrum in CDCl3 of P1; (b) 1H-NMR spectrum in CDCl3 of P2. | ||
Generally, the coupling of 3-substituted thiophenes via 2- and 5-position leads to polymers with various degrees of regioregularity due to the formation of head-to-head (H–H), head-to-tail (H–T) and tail-to-tail (T–T) configurational isomers49 and the complexity of 1H-NMR spectra of such polymers results from the presence of four triads of monomeric units (HT–HT; TT–HT; HT–HH; TT–HH) that would create slightly different environments for the protons in the central rings.49b,d,e
The reported data in the literature showed that, by using different NMR analysis and also based on model configurational triads,49d it was possible the assignment of the protons in the 4-position of the ring in all the triads. In the spectrum of polymer P1 four groups of peaks can be identified in the aromatic region. The protons of type g1 can be found in TT–HT and TT–HH triads and those marked in Fig. 1a as type g2 could be attributed to the thiophene proton in triads of type HT–HT, TT–HT and HT–HH.49d
The observed spectrum is consistent with a regiorandom poly-3-substituted thiophene for which (based on NMR integration) the triads are present in the following ratio: HT–HT (δ 7.37–7.47 ppm)/TT–HT (δ 7.48–7.72 ppm)/HT–HH (δ 7.75–8.02 ppm)/TT–HH (δ 8.23–8.26 ppm) = 8%/41%/32%/19%.
Additional configurational information can be abstracted as well from the intensity ratio of the two peaks centred at 4.3 ppm and 4.5 ppm attributed to the protons marked a in Fig. 1a. Previous reports showed that, due to the difference in configuration, α(1)-methylene protons and even β(2)-methylene protons in poly-3-alkylthiophenes present splitted peaks associated with HH (δ = 2.56 ppm) and HT (δ = 2.79 ppm) diads.49b,c,e From the intensity ratio of the two peaks at 4.3 ppm and 4.5 ppm it can be assumed that the HT diads are present in polymer P1 in 66% ratio whereas HH diads are in 37%. It can be also concluded that, in the case of M1, this type of Yamamoto reaction shows regioselectivity in the favor of HT configurational isomers. Moreover, in the spectrum of P1, weak peaks (i, j, k, g3) are also discernible. Former 1H-NMR studies concerning to regioregularity of various poly (3-alkylthiophene-2,5-diyl)49a,e shows that such peaks should be attributable to the protons of the thiophene terminal structural units. In our case, the singlet at 7.04 ppm and the broad signal at 8.30 ppm could be assigned to 2-position terminal protons and 5-position terminal protons i.e. protons j and i, respectively.
The terminal proton in the 4-position of thienyl ring will have two chemical shifts (k and g3 in Fig. 1a) depending on whether the terminal ring is connected to the main chain by the 2- or 5-position, respectively. While for the protons labeled g3 a shallow doublet is present in the range 7.17 –7.20 ppm, the proton labeled k is shifted downfield compared to proton g3 and is, therefore, masked by the large signals due to the protons located on non-terminal thienyl rings, in a similar manner as observed for poly (3-alkylthiophene-2,5-diyl).49a
In the 1H-NMR spectrum of P2 the aromatic peaks are shifted to lower δ ppm presumably due to the presence of CH2 groups in the vicinity of thiophene rings. So, the aromatic signals of P2 are partially covered by the signal of CDCl3 and similar calculations as those performed in the case of P1 could not be done. However, the broadening of the peaks from the aromatic region indicates the formation of the polythiophene sequence.
The third macromonomer, 5T in Scheme 1, was synthesized via a Suzuki coupling reaction between M1 and 2,2′-bithiophene-5-boronic acid pinacol ester and details about its synthesis and structural characterization were already reported.43b
Photophysical properties of bare pentathiophene reported in the literature have been interpreted as indicating that this compound acts as the first representative of an “α-polythiophene”.50 Therefore, we considered interesting to study the 5T photophysical properties in aqueous media along with its self-assembling in solution, cytotoxicity and cell imaging capability and to compare the results with those of P1 and P2. Moreover these results could be very useful, more so as recently very interesting biomedical applications were reported for different pentathiophenes decorated with ionic functionalities.30,31b
In the next step, as is shown in Scheme 1, following an already reported method,37a the macromonomer 5T was polymerized by oxidative coupling in the presence of anhydrous FeCl3. In our previous reports 5T was polymerized anodically and the formed polymeric films were investigated in terms of morphology and topography.35d,43b Meanwhile it was shown that the obtained films could work as active surfaces for the selective adsorption of proteins,43b concomitantly being excellent cellular matrices and promising materials for biotechnological applications in which electrochemical properties are used as elements for the communication with cells.35d It is well known that different polymerisation methods could conduct to materials with different properties and, in this context, in contrast with the polymer obtained by electrochemical polymerisation of 5T,43b P5T was expected to be an water soluble polymer that self-assemble in fluorescent micellar nanoparticles in aqueous media.
In the current attempt P5T resulted as a reddish powder after its treatment with hydrazine-hydrate for dedoping to a neutral product and its 1H-NMR spectrum (Fig. 2b), shows, as expected, broad peaks in the aromatic region as a result of the polymerization process. In the same figure the 1H-NMR spectrum of 5T is enclosed for comparison.
 | ||
| Fig. 2 (a) 1H-NMR spectrum of 5T in acetone-d6; (b)- 1H-NMR spectrum of P5T in acetone-d6. | ||
IR spectra of macromonomers M1, M2 and 5T and those of the corresponding polymers P1, P2 and P5T are presented in Fig. S4 (ESI) and detailed discussed in ESI.† The presented results complete those obtained by 1H-NMR characterization and all together confirm the proposed structure of the compounds reported in this paper.
The apparent molecular weight of macromonomers and derived polymers were measured by GPC using THF as solvent. The obtained GPC traces are presented in Fig. S3 (ESI†) and the values are given in Table S1, (ESI†). Due to the reasons that were detailed in ESI part,† these values must be considered with care, and theoretical values for the molecular weights of the macromonomers were used for the recipe calculations.
The polymers thermal behaviour was followed by TGA, DSC and POM investigations and the experimental data regarding the thermal stability and melting temperatures are presented in Table 1. Also the values of the similar parameters for PEG2000 were included for comparison. Concerning the thermal degradation of the synthesized polymers, it can be observed that it is a combination between thermal degradation of the PEG2000 and that of bare poly (thiophene-2,5-diyl).
| Polymer | IDTa (°C) | Twmaxa (°C) | YC600a (%) | Tmb (°C) |
|---|---|---|---|---|
| a Data determined from TGA measurements: IDT-initial degradation temperature; Twmax-the temperature at which the weight loss is maxim; YC600-the percent of char yield at 600 °C.b Data obtained from DSC measurements: Tm-melting temperature; h1-first heating; h2-second heating. | ||||
| PEG2000 | 311.7 | 441 | 5.00 | 54.4 |
| P1 | 319 | 410 | 15.36 | 53.32(h1); 52.32(h2) |
| P2 | 336 | 406 | 16.97 | 53.24(h1); 52.83(h2) |
| 5T | 250; 322 | 406 | 12.11 | 53.08(h1); 52.35(h2) |
| P5T | 229; 324 | 406 | 10.42 | 50.47(h1); 50.02(h2) |
It was reported that the thermal degradation of the unsubstituted polythiophene under nitrogen is a three steps process,51 with an initial degradation temperature around 260 °C, showing a weight loss of 7% at 380 °C and a remarkable char yield of more than 50% at 900 °C.51b
The thermal degradation of PEG2000 is one-step process, which starts at 312 °C with a weight loss of 97% at 440 °C.
Polymers P1 and P2 that contain a PEG side chain in each structural unit, similarly as PEG2000, degraded in only one step, showing initial degradation temperatures (IDTs) slightly higher than PEG2000, concomitantly with approximately three times higher YC600 (Table 1).
The weight loss of around 85% registered for P1 and P2 at 600 °C could be attributed to the degradation of the whole polymer.52 A sharp weight loss attributable to the thermal cleavage of the carboxylic ester moiety, which has been observed in other cases53 wasn't involved in thermal degradation of P1 and P2, respectively.
The thermal degradation of quinquethiophene 5T and its derived polymer P5T occurred in two steps, the weight loss of the first step being 3.76% and 5.1%, respectively.
Taking in account the lower PEG side chains density for these compounds by comparing with P1 and P2, a higher similarity with the thermal degradation of poly (thiophene-2,5-diyl) is expected for P5T and 5T. Thus, their first weight loss could be attributed to the first degradation step of oligo/polythiophene main chains, as for poly (thiophene-2,5-diyl).51 The second weight loss could be due to the degradation of the ester linkages and of PEG side chains, as well as to the completion of oligo/polythiophene main chains degradation. Char yields higher than that of PEG2000 resulted also for 5T and P5T.
The synthesized P1, P2, 5T and P5T contain in their structures oligo/poly(thiophene-2,5-diyl) main chains that were reported as crystalline or semicrystalline materials49b,51,54 and also crystalline PEG as side chains. As can be observed from the data displayed in Table 1 and in Fig. S5 (ESI†), the DSC measurements of all materials performed in the range 35–250 °C (for P5T up to 200 °C) revealed only one endotherm. These thermal phenomena are attributable to the melting of the PEG side chains. The values of the determined melting temperatures (Tm) are only slightly different from the melting temperature of PEG2000, higher difference being registered for 5T and P5T. Even if α-quinquethiophene was reported as having a melting point at 256 °C,54c no other melting phenomenon was observed for our 5T in the investigated range. The slightly different values registered for (Tm) of all investigated polymers in the first heating and the second heating, respectively (Table 1), could derive rather due to the different type of PEG crystallization than due to the different heating rate used for measurements (see details in caption of Fig. S5, ESI†). This assumption could be true taking in account that in the first heating are investigated polymeric materials as they resulted by precipitation from their solutions and in the second heating is followed the behaviour of the polymers as they resulted after the controlled cooling from their melts.
When a decreased heating rate as 3 °C min−1 was used for P1 investigation in a cycle “heating–cooling–heating” (Fig. 3 A), two melting endotherms in the first heating, one crystallization exotherm in the cooling and only one melting endotherm were observed.
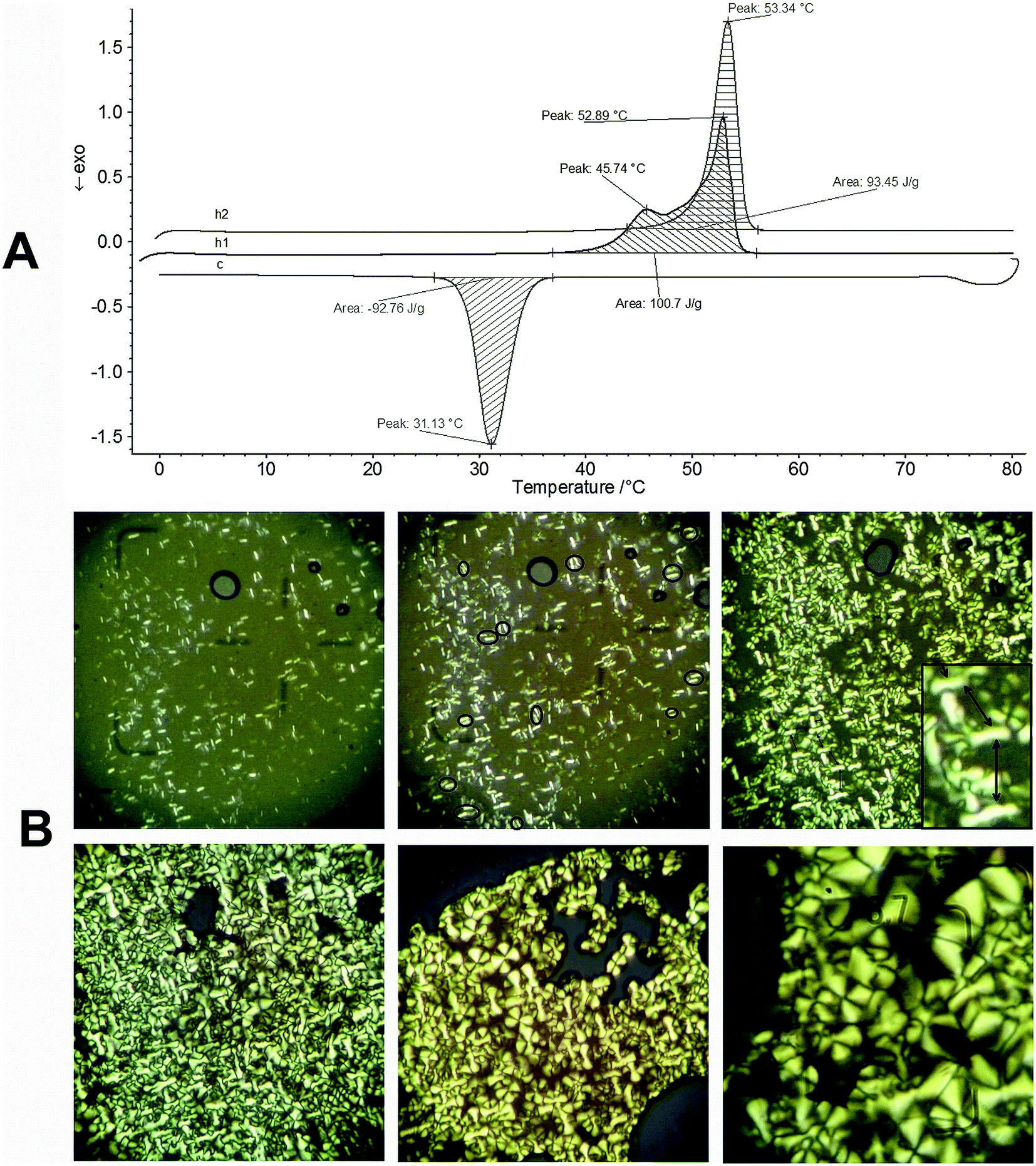 | ||
| Fig. 3 (A) DSC thermograms for polymer P1 registered at 3 °C min−1, h1-first heating, c-first cooling, h2-second heating; (B) optical micrographs of P1 under polarized light, in the second cooling, from 54 °C to room temperature, at a cooling rate of 2 °C min.(the photos show the cooling cycle from left to right and from up to down). | ||
The double melting peaks of PEG were also observed by other scientists55 and were attributed to the presence of the PEG crystalline lamellae with different fold numbers. In the second heating run only one peak around 53 °C was observed, inducing the conclusion that the rate of 3 °C min−1 in the cooling run was slow enough to permit an uniform crystallization of the all PEG side chains. The X-ray diffraction of the polymers in powder form was also investigated and the obtained XRD profiles are presented in Fig. S6, (ESI†). In the literature, X-ray structural studies of non-substituted thiophene oligomers54a–c and poly(thiophene-2,5-diyl),49b,51 showed a herringbone-type molecular packing, while poly(3-alkylthiophene-2,5-diyl) support a lamellar structure in which parallel stacks of polymer main chains are separated by regions filled by the alkyl side chains.49c,e Consequently, while in the diffractograms of the thiophene oligomers and poly (thiophene-2,5-diyl) three main X-ray diffraction bands are present in the 19–29° 2θ region,49b,54b in the XRD profiles of poly(3-alkylthiophene-2,5-diyl), especially in those of the regioregular ones, the characteristic peaks appeared at around 2θ angle 5.4°, 10.8°, 16.5° and 23.5°.49c,e
XRD diffractograms of the thiophene oligo/polymers reported in this study have almost similar shapes to each other and, as expected, they are dominated as in the case of the IR spectra by the peaks belonging to the PEG side chains at 2θ angles 13.5°, 15°, 19°, 23°, 27°, 32° and 36°. If present, some characteristic peaks of poly (thiophene-2,5-diyl) main chains, in a crystalline organization, were merged together with those of PEG in the 2θ region of 19–24°. In Fig. S6, (ESI†), several exceptions with low intensity are marked at 6.4° and at 9.1° for P1, at 5° for 5T and the most obvious one for P5T appeared at 29.3°. Position of the latter peak is similar with the third peak position in thiophene oligomers54a–c and in poly (thiophene-2,5-diyl).49b,51 The presence of this peak only in the diffractogram of P5T suggest that the density of the PEG side chains on polythiophene main chains could influence in bulk molecular packing of the investigated polymers.
If in the X-ray diffraction study the ordering character of the polymer chains in a microscopic scale can be assessed, POM investigation was used to give an outline of in melt properties of P1 in a relatively macroscopic scale. The yellow powder of polymer P1 was run between room temperature and 54 °C for two times in a cycle “heating–cooling”, using a heating rate of 2 °C min−1. In the first heating, at 47 °C, under pressing, the material is birefringent in a fluid-viscose state and, at 53 °C in the first heating and at 54 °C in the second one, the material become isotropic. The images in Fig. 3B present the evolution of the material during the second cooling from 54 °C to room temperature. In the first image (top row, left side), short, bright rods spreaded into a compact, isotropic melt can be seen. If the definition given by Wegner a decade ago for the “hairy-rods” is taken into account, (in the solid and solvent-free state the “hairy-rods” are “molecular composites”, side-chains giving rise to a continuous matrix in which the rod backbones are embedded as reinforcing elements),23 the first image could be an appropriate visual representation of this definition. The experimentally observed rods of P1 could be formed in melt from bundles of polythiophene main chains most probably due to the π–π stacking phenomenon. Assemblies of two such rods can be seen very often in the second image (marked with circles), while in the third image, concomitantly with the PEG spherulites appearance, the rods were moved away one another (black arrows). The PEG spherulites in a “classical” shape55 appeared in both cooling cycles starting from 40 °C and they have developed and increased in size as long as the temperature decrease toward room temperature, while the rods became trapped within the interfibrillar regions of the growing spherulites.56 A continuous film with stable morphology was obtained by crystallization from isotropic state, no cracks being present along days. The temperature values for melting and crystallization of P1 determined by POM investigation were almost similar with those obtained by DSC (Fig. 3A), the small differences being derived most probably from the different heating–cooling rates.
Self-assembly properties in aqueous solutions
Because of their amphiphilic nature, P1, P2, 5T and P5T are soluble in a wide range of non-polar and polar solvents such as chloroform, benzene, methanol, dioxane (as a few examples), being insoluble in cold diethyl ether. Also they are forming micellar nanoparticles in water and in the buffer saline solutions.The fluorescent micellar nanoparticles of our synthesized materials in aqueous media were achieved by direct dissolution method. This method was chosen based on already mentioned reason of the calculated value of the HLB; moreover, it was demonstrated that in the case of amphiphiles with special architecture, stable micelles can be also obtained below the typical values of HLB.57 For most molecular amphiphiles there is a critical micellar concentration (CMC) below which these amphiphiles dissolve in unimolecular form. Typical CMC values of polymeric surfactants range between 10−9 M and 10−4 M.39b However it was already emphasized in the literature that some amphiphiles have no CMC.39b Several explanations have been reported for a such behaviour: (i) the association concentration of the amphiphiles is extremely low and beyond of any device ability to detect it or (ii) the dissolution of the amphiphiles samples occur by a process involving fragmentation of the powder grains into smaller parts until colloidal dimensions are reached.58
It is also the case of our investigated polythiophenic materials that resulted in the aggregated micelles at the concentration of 5 × 10−10 M (Table 2). A similar behaviour was also recently reported for some conjugated polyelectrolytes.59
| Sample | Diametera (nm) in water | Diametera (nm) in buffer saline | Diameterb (nm) in dry state |
|---|---|---|---|
| a Determined by DLS at concentration of 5 × 10−4 μM based on repeating unit (RU).b Determined by AFM on films from water dispersions; The diameters were taken from the width of the peak at the baseline in sectional height profiles. | |||
| P1 | 271 | 60 | 60 |
| P2 | 254 | 111 | 80 |
| 5T | 265 | 45.6 | 100 |
| P5T | 149 | 59.2; 252 | 200 |
To prove the self-assembling of the amphiphilic synthesized polythiophenes in water and to characterize the formed micellar nanoparticles in terms of size and shape, 1H-NMR spectroscopy, DLS, SEM and AFM techniques were complementary used. While the 1H-NMR highlights the presence of the nanoparticles and DLS allow for their dimensional analysis in the solvated state, the microscopic investigations (SEM and AFM) give an image of their shape and size in dried state, even if the PEG corona in dried state is collapsed and consequently in a different conformation.60
It is well established that NMR is an important tool for detecting the formation and structures of micelles in solutions of diblock copolymers.61 For polymer P1, 1H-NMR spectrum in D2O, that is given in Fig. S7 (ESI†), revealed only the characteristic peaks of PEG side chains, whereas the polythiophene main chain peaks are collapsed, implying that their molecular motion in water is restricted. These 1H-NMR experimental data suggests that the polythiophene main chains form a central solid-like hydrophobic core to minimize their interaction with D2O, while the PEG side chains interact favourably with water molecules forming hydrogen bonds which stabilizes core–shell type particles structure of this “hairy rod” polymer. By using the same technique an analogous behaviour was also pointed out for other PEG-containing graft copolythiophenes.62
For the nanoparticles possessing PEG side chains it was reported that PEG forms a flexible layer on their surface.60 When the density of the PEG side chains is high, these chains start to interact each other and the excluded volume effect forces them to extend into a brush-like configuration. When the surface PEG density is relatively low it forms a mushroom-like structure to maximize surface coverage.63 The hydrogen bonds between the oxygen ether linkages in structural units of PEG and water molecules lead to the formation of the “structured water” around their flexible polymer chain.
The hydration number of a PEG molecule depends on its configuration. It was reported that in the brush configuration PEG is more accessible to water, binding approximately 30% more water than PEG in the mushroom regime. For example by using DSC analysis it was shown that each molecule of PEG having the molecular weight of 2000 (PEG2000) binds 136 molecules of water while for PEG2000 covalently attached to the lipid molecules organized in micelles the water binding increase to 210 molecules.63
Based on these assumptions the DLS obtained results for the investigated polymers, presented in Table 2 and Fig. S8, (ESI†), can be explained as follows: (i) the size of the nanoparticles of P1 and P2 in water are in the same range, most probable due to the similar density of the PEG side chains on the surface of the nanoparticles (an influence of the molecular weight on the nanoparticles size in water could also be expected);64 (ii) the lower value obtained for the polymer P5T in water, by comparing with P1 and P2, could be due to the lower density of the PEG side chains present on the surface of the formed micelles which consequently will conduct to a lower amount of molecules in “structured water”, namely to a thinner shell.
The nanoparticles size, obtained from the AFM investigation on polymers films obtained from theirs water dispersions on mica substrate (Fig. 4 and 5), emphasized a higher value for polymer P2 nanoparticles by comparing with polymer P1. It can be concluded that when the amount of the “structured water”, which was eliminated during the film preparation by freeze-drying, is the highest then the size of the nanoparticles in the dried state is the lowest. In the case of P5T, based on the same reasoning, the highest size of the nanoparticles in the dry state was expected due the lower amount of the “structured water”. As can be seen in Table 2, indeed the highest value was registered for P5T but this value is much higher than that obtained by DLS measurement in water dispersion. This phenomenon could be explained based on two assumptions. One of them is based on the already reported tendency of PEG to crystallize during freezing, crystallization that conducts to intra- and inter-particles bridges of crystallized PEG, namely to aggregated particles after water removal.65 But this argument is less probable taking in account that similar phenomenon wasn't observed for P1 or P2. Particles aggregation during freeze draying of P5T could be due more probable to the lowest PEG density on the surface of the nanoparticles, which may favour also their core–core interaction (Fig. 4).
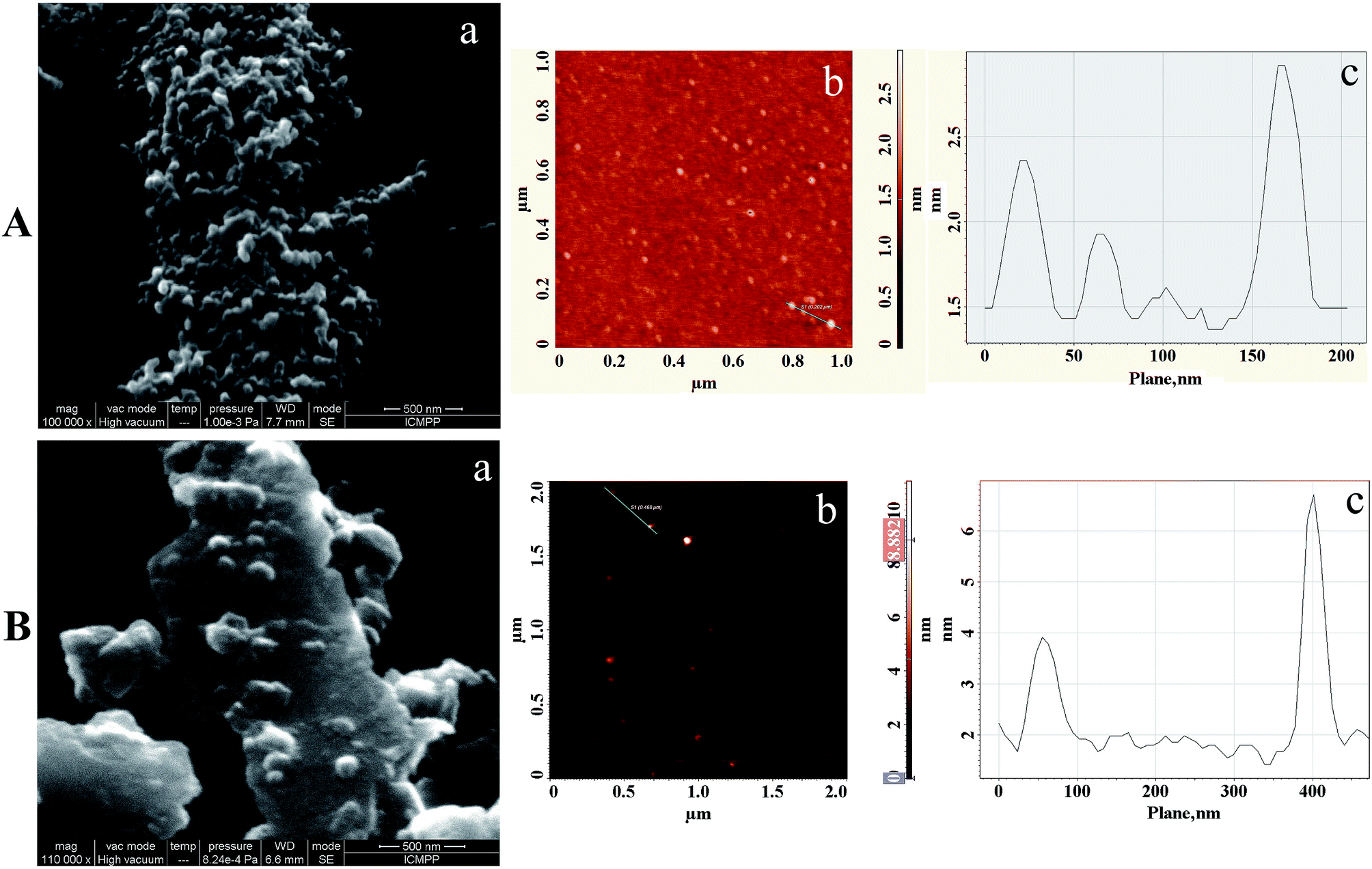 | ||
| Fig. 4 (A) SEM (a), AFM image (tapping-mode) (b) and cross-sectional analysis (c) of P1 micellar nanoparticles; (B) SEM (a), AFM image (tapping-mode) (b) and cross-sectional analysis (c) of P2 micellar nanoparticles. | ||
A special behavior has to be noticed for the quinquethiophene 5T observed by both DLS and AFM measurements. In the case of its structure the calculated HLB has the lowest value (ESI Table S1†) and, to form stable nanoparticles based on hydrophobic interactions in the core, more oligothiophene chains are required to self-assembly, thereby accounting for the increased size. This could be the reason for which the nanoparticles size in water for 5T is comparable with those of P1 and P2 and becomes higher then these in the dry state based on above-mentioned reasoning for P5T (Fig. 5).
 | ||
| Fig. 5 (A) SEM (a), AFM image (tapping-mode) (b) and cross-sectional analysis (c) of 5T micellar nanoparticles; (B) SEM (a), AFM image (tapping-mode) (b) and cross-sectional analysis (c) of P5T micellar nanoparticles. | ||
It is also noteworthy that the self-assembly can also occur at solution/substrate interface during the drying process.66 In this case some of the seen structures in the SEM and AFM images could be formed during the solvent evaporation, they being present in the obtained films near to the nanoparticles already formed in solution and deposited on the substrate. Moreover, the interactions between nanoparticles on the one hand and nanoparticles and the substrate on the other hand, which may occur during the film formation, should be taken into account, too. The affinity of the PEG shells for the hydrophilic mica substrate results in a layered morphology of the films with embedded nanostructures that can be seen in both SEM and AFM images. Generally, the apparent diameter of the nanoparticles in Table 2 is 2–4 times larger when measured using DLS than when measured from AFM. Similar observations were reported in literature.64
As the hydrodynamic diameter (DH) is a good indicator of the particle size in vivo, its measurement was also performed in phosphate buffer saline at similar concentration as that used for in water measurements. From the data presented in Table 2 smaller dimensions are found by comparing with those obtained in water. The explanations could be as follow: (i) the complexation of the inorganic cations of the salts present in the medium disrupted the hydrogen bonds between PEG chains and water, thereby partially dehydrating them. The repulsion due to the brush regime is thus reduced, therefore the interfacial tension is also reduced resulting in smaller size of micellar nanoparticles (ii) the pendant PEG chains are required to distort from a chain-extended conformation to accommodate the larger cations and to form a multidentate complex,67 phenomenon which also could conduct to a decreasing in the nanoparticles size. It can be also seen that in the micellar solution of P5T in phosphate buffer saline there are two types of particles as size. It can be supposed that the smaller particles could result from the aggregation of few macromolecules, while the larger objects formation could be due to the lowest PEG side chains density which can favours the core–core interactions between already formed small nanoparticles.68
The dimensions of the nanoparticles of investigated polymers were found to be in the proper range to avoid fast renal clearance (DH > 10 nm) and still below the cut-off size of the leaky pathological microvasculature of hypervascular tumors (DH < 200 nm),69 thus making them candidates for application in fluorescence imaging based on the EPR effect. This assumption is based on the fact that repeated measurements of the particle size in solutions stored over several months in the dark resulted in the similar data as for the initial measurements.
Optical properties in aqueous solutions
The photophysical properties of the studied polymers were followed in water by UV-Vis and fluorescence measurements, and the results are presented in Table 3, Fig. 6 and Fig. S9 (ESI†). For a realistic interpretation of the obtained data, a survey of the literature44e,70 concerning the optical properties of different poly-3-PEG-substituted thiophenes in solution was done (see also the references in Table S2, ESI†), which conducted to the conclusion that the absorption and emission maxima in good solvents for both backbones and PEG side chains depend, at least, on the following factors: (i) the number of PEG structural units in the side chains; (ii) the modality of connection of PEG side chains to the 3-position of the thiophene rings, namely the connection through a methylene bridge or through an oxygen atom; (iii) the used method for the polymerization of PEG-containing thiophene monomers; (iv) the presence of an additional substitute in the 4-position of the thiophene ring.| Samples | λmax absb (nm) | λmax emc (nm) | Stokes shift (cm−1) | εd (M−1 cm−1) | ΦFe (%) |
|---|---|---|---|---|---|
| a The concentration of polymer solutions used for both type of measurements were of 5 × 10−4 μM based on repeating unit (RU); The bold, italicized numbers represent the wavelengths of band maxima, the other values being present in the spectra as shoulders.b The values in the square brackets correspond to the values of λmax abs of polymers solution in PBS (25 mM; pH = 7.4) at the same concentration as in water.c λex = λmax abs; The values in the square brackets correspond to the values of λmax em of polymers solution in PBS at the same concentration as in water.d Molar absorption coefficients calculated at the main λmax abs value.e Absolute fluorescence quantum yield of the solutions in water at concentration of 5 × 10−4 μM RU. | |||||
| P1 | 400; 505; 520; [402] | 527; 564; 594; 631; [538] | 5952 | 53500 |
2.31 ± 0.01 |
| P2 | 363; 505; 520; [310] | 467; 517; [444] | — | — | — |
| 5T | 370;467; 505; 520; [372] | 504; [513] | 7186 | 93800 |
30.63 ± 0.057 |
| P5T | 400; 469; 505; 520; [403] | 484; 488; 508; 594; 612; [490] | 4508 | 47500 |
1.15 ± 0.008 |
 | ||
| Fig. 6 (A) Fluorescence spectra of micellar nanoparticles of investigated materials dispersed in water; the photo in the inset represent the water dispersion of polymer P1, oligomer 5T and polymer P5T in the day light (bottom line) and under excitation with a 365 nm UV lamp (top line); (B) photostability of fluorescent micelles of P1, oligomer 5T and polymer P5T in water dispersions by irradiation with a mercury lamp. The materials used concentration was 5 × 10−4 μM based on repeating unit (RU). | ||
Thus, when the reported data on polythiophenes with PEG side chains containing two, three, four, or seven repeating units (RU) attached at the 3-position of the thiophene ring are taken into account, it can be concluded that the values at which the maximum of absorption and that of emission appeared decrease with the increase of the PEG chain length.
Also, when the connection between thiophene rings and the PEG side chains is mediated by an oxygen atom, these values are higher than those for which the connection was realized through a methylene bridge and the blue shift increase with the number of methylene moieties in these bridges (ESI, Table S2†).
These experimentally observed phenomena seem to be the result of concerted factors. At first glance it seems that in good solvents for both main and side chains, the increase of the PEG side chains bulkiness and any modification of their organization could be the driving force of the optical effects and hypsochromically shifted absorption and emission maxima can be attributed somewhat speculatively to the increased steric hindrance of the side chains which forces the polythiophenes backbone to adopt a non-planar configuration.
Also it is worthy to mention the already reported observation that the use of an ester linkage to attach an alkyl substitute at the 3-position of a thiophene ring induced a blue shift for absorption and emission of the resulted polymers by comparing with those for which the alkyl substitute was directly connected to the ring.71
In the context of the above presented results, the blue shifted values of λmax abs and λmax em for polymers P1, P2, P5T and oligomer 5T (Table 3) in comparison with those presented in Table S2 (ESI†) could be attributed to the presence of the long PEG side chains of 44 RU and also to their connection at the 3-position of the thiophene ring through an ester linkage. Moreover, when λmax abs and λmax em values of P1 and P2 are compared, a significant blue shift of these optical characteristics can be observed for P2, too (Table 3 and ESI Fig. S9†). These differences observed for polymer P2 relative to P1, could be due to the presence of the methylene bridge between the thiophene ring and ester-ended PEG2000 substitute (see Scheme 1), which disrupts the conjugation between thiophene chain and carboxyl moiety of the ester, having as a result not only the blue-shifting of absorption and emission maxima but also a very pronounced decrease of the fluorescence intensity (Fig. 6A). Polymers P1 and P5T show the main absorption maxima at the same wavelength (see Table 3 and ESI Fig. S9A†).
It means that the density of the PEG side chains to the backbone seemingly did not influence this parameter.
All above comparisons of our polymers were related to the reported poly-3-PEG-substituted thiophenes with short PEG side chains in a common good solvent for both main chains and side chains. As long as the interpretation of molecular mechanism responsible for spectroscopic changes in water soluble polythiophenes resulted in often controversial and contradictory interpretation, (due to the difficulty of distinguishing conformational changes of individual molecules from intermolecular interactions),28a,c the assignment of our experimentally obtained data can be more reliable if compared with those of others polythiophenes, equipped with longer or shorter PEG moieties, investigated in water. Unfortunately such data are scarcely reported in the literature.70a,72 It seems that PEG chains containing 7 or less number of RU does not induce water solubility with a few exceptions, (ESI Table S2†); all existing results on this subject suggest the key role of water as poor solvent for the main chains in organizing the polythiophenes by solvophobic interactions conducting to different secondary structures as aggregates70a or foldamers.72b The authors attributed the presence of the two or more red-shifted peaks of such polymers, in both the absorption and emission spectra in water, to the concomitantly presence of non-planar/planar conformational structures of the polythiophenic backbones. The wavelengths values at which these maxima appeared in water followed the same trend as for polymers solutions in good solvents, namely the longer the PEG side chains the shorter the wavelengths at which the absorption or emission appeared (ESI Table S2†). Therefore, as our synthesized polymers contain longer PEG side chains, the blue shifted values in Table 3 relative to reported poly-3-PEG-substituted thiophenes would be expectable.
In our case, the experimentally observed optical properties are the result of a more complex and concerted phenomena related to the peculiar molecular architecture of the synthesized “hairy rods” which differentiate them from the usual graft copolymers.20b
Recently it was pointed out that “molecular brushes” can be regarded as molecular tensile machines. The significant, controlled self-generated mechanical tension along the backbone is one unique feature of such architecture, which directly impacts the electronic structure reflected in photophysical properties when the backbone is a polythiophene.73a
Moreover, the polymers aggregation within the micellar nanoparticles cans be affected by the spatial spherical constraint of the micelle core, which in turn influences the photophysical properties.73b
As can be seen in Fig. 6A and in Fig. S9(A), (ESI†), for our synthesized polymers, both absorption and emission spectra are asymmetric in shape, with vibronic fine structure attributable to the nanoparticles formation. The red shifted shoulders relative to the main peaks and long red tails appeared in both absorption and emission spectra. They are similar to those often observed in thin films,74 which have been assigned to the occurrence of the interchains energy transfer that is possible in the solid state.70b We have to point out the presence in the UV-Vis absorption spectra of all polymers investigated in this study of the shoulders at similar values of 505 nm and 520 nm. This phenomenon can be interpreted as a mark for their similar “hairy-rod” architecture, for which the repulsion among the long PEG side chains leads to the presence in some extents, in the nanoparticles structure, of more rigid and extended conjugated polythiophenes backbones of similar length that show red-shifted absorptions. In spite of the steric effect, these more planarized conformations exhibit even stronger interchains interaction than their twisted counterparts,28c and the presence of the red tails in fluorescence spectra could be related to the fraction of such red-shifted aggregate species in the nanoparticles structure.
It is also interesting to notice that in the case of oligomer 5T, when comparing its optical characteristics in Table 3 with those reported for unsubstituted α- pentathiophene,50 a blue shift of λmax abs of 47 nm concomitantly with a bathocromically shifted λmax em with a magnitude of 22 nm were observed. Also a more than two times increase of molar absorption coefficient (ε) for 5T and almost the same value for ΦF (0.32 for α-pentathiophene measured in ethanol;50 0.31 for 5T in water, Table 3) can be remarked. Based on all of these observation it can be concluded that in the case of 5T the λmax abs blue-shifting is most probable due to the presence of the ester function which connect the PEG side chains to the main chain and not to conformational reasons. An optimal balance between hydrophobicity/hydrophilicity and rod–coil ratio, as well as the peculiar architecture of this amphiphile could be accountable for such behavior. As it was already pointed out,8c,28c it seems that the presence of PEG shell around the π-conjugated system of the nanoparticles of 5T provide an electronic shielding from the external environment, conserving its optical properties. Taking in account this observation, the fluorescence behavior of polymers P1 and P5T can be similarly ascribed to their structural and architectural features. Therefore, the higher density of the PEG side chains can induce a higher steric hindrance of them in the case of P1 as compared to P5T, phenomenon which can leads to a more rigid and extended conjugated backbones for P1 than for P5T. The RU of P5T is characterized by a ratio of PEG2000 side chain to thiophene rings of 1/5 while for P1 this ratio is 1/1. Due to this lower ratio, the number of the polymeric chains necessary to form a stable micellar nanoparticle is higher for P5T than for P1, as was already confirmed by DLS and AFM analyses. In this case the chains packing defects, resulted as a consequence of the stress imposed by the spherical confinement of the micellar nanoparticles core, could be increased, this fact having as result a blue-shifted emission of P5T relative to P1 and also the lowest values of ε and ΦF (Table 3).
A similar trend can be observed when P5T is compared with 5T. It seems that the 5T main chain length is an optimal one, which best fits to the spherical confinement of the micellar nanoparticles core in comparison with the longer P5T main chains. The polythiophene main chains form a central solid-like hydrophobic core to minimize their interaction with water. As long as in the solid state the thiophene ring is an easily deformable heterocycle, which adapt its geometry to the environment,54c the more kinked or twisted main chains of P5T that fit to the geometry of the micellar nanoparticles core would be expected. In the case of P5T all of these aspects conducted to the most blue-shifted λmax em and to a blue fluorescence of its water solution, as can be seen in the inset of Fig. 6A.
If λmax em of P1 and P5T nanoparticles are compared with that of the unsubstituted polythiophene in thin film (λmax em = 545 nm),75 both of them are blue-shifted, but this blue shift is limited, especially for P1. This means that the polythiophene chains in the micellar nanoparticles core can be slightly twisted or folded in a more or less extent as comparing with those forming the 2D geometry of the thin film. On the other hand, nanoparticles of unsubstituted polythiophene (PThN) were recently prepared by oxidative polymerization in aqueous medium in the presence of a surfactant, and their photophysical properties in aqueous emulsion were evaluated.41b,c While for all the size of PThN broad UV-Vis spectra with λmax abs centered at 400 nm were observed, for photoluminescence property a size-dependent effect was emphasized,41c namely the highest value of λmax em (511 nm) was registered for the highest size (51 nm) of PThN. Meanwhile a decrease of fluorescence intensity with the increase of the nanoparticle size was also pointed-out. The authors' explanation for such behavior was based on the variation of the total emitting area with size variation and on the self-absorption effect.41c By comparing the values of λmax em for P1 and P5T given in Table 3 with those reported for PThN, it can be observed that they vary almost in the same range, with a red shift for P1. From the both above comparisons (with polythiophene thin film and with PThN) it seems that PEG shell of the studied micellar nanoparticles protect and preserve the fluorescence property of the polythiophene backbones in the core, and that the PEG side chains chemically connected to them not exert an effective negative influence on the main chains conformations. Moreover, if the fluorescence intensity of P1, 5T and P5T in Fig. 6A and the red-shifting of λmax em in Fig. S9(B) (ESI†) are analyzed in conjunction with the values of micellar nanoparticles size presented in Table 2, a similar size-dependent fluorescence as for PThN could be emphasized. However at this stage this is only a speculative observation, a systematically and deeper study necessary to exactly establish the mechanism for the experimentally observed phenomena being on the way. The values of molar absorption coefficients (ε) of P1, P5T and 5T in Table 3 are higher than31a or in line50 with those observed for other polythiophenes in solution. Absolute fluorescence quantum yields (ΦF) of P1 and P5T are in the range usually observed for polythiophenes in the film state.71 As long as the brightness directly depends on ΦF and ε,76 the high values of ε can compensate for lower ΦF,77 and the fluorescence brightness of nanoparticles remains high enough to be used for fluorescence imaging applications. Moreover it is know that the viscous cellular environment can affect the fluorescence in an unpredictable way,31a so the values in Table 3 can indicate only a possible trend of the investigated materials within the cells.
The photostability of the fluorescent nanoparticles is particularly important when fluorescence-based imaging techniques, especially long-term imaging or tracking experiments are considered. We tested the photostability of P1, 5T and P5T by placing their water solutions under irradiation of a high-pressure mercury arc lamp, at 365 nm, with an intensity of 1.1 mW cm−2 and the fluorescence spectra were measured at time intervals. As can be seen in Fig. 6B, even if 5T lost 20% of its initial fluorescence intensity, all investigated materials show remarkable photostability after 120 minutes of irradiation, thus meeting one of the most important requirements if their application in cell imaging is intending. Having also sufficiently high absorption coefficients, large Stokes shifts to filter out the exciting light, obvious emission tail in the wavelength region >600 nm as is showed in Table 3 and bright fluorescence (inset of Fig. 6A), 5T, P1, and P5T could serve as a good fluorescent molecules for optical imaging. Following this line, as cellular studies are normally performed in PBS, we collected the absorption and fluorescence spectra of our compounds in 25 mM solution of PBS and the values of the λmax abs and λmax em are presented in Table 3. By analyzing these values in comparison with those in water it can be revealed that, with the exception of P2, all the other compounds showed a slight red shifting for both parameters. Concerning polymer P2 it was shown once more that the presence of methylene bridge between the thiophene ring and ester-ended PEG side chains constitutes a flexion point for these side chains that affects all the optical properties in a different manner as for the other investigated compounds.
It was also investigated the effect of non-specific interactions on the polymers fluorescence that can take place in both culture medium and cells environments due to the presence of a variety of biomolecules. In this aim BSA was chosen as an ideal model, being abundant in culture medium and the most abundant in blood plasma protein in mammals.78 Changes of fluorescence spectra for some of investigated polymers upon the addition of BSA with concentrations ranging from 0 μM to 0.25 μM in steps of 0.05 μM in 25 mM PBS were monitored. It was observed that after a slight change in fluorescence intensity the saturation occurs at BSA concentration of 0.25 μM as shown in Fig. S10 (ESI†). This stability of fluorescence could be due to the presence of the bio-repellent PEG side chains forming the shell of the investigated micellar nanoparticles.
Preliminary results of “in vitro” fluorescence non-specific cellular imaging
To evaluate the potential of nanoparticles developed in this work as fluorescent biomarkers, cytotoxicity investigations and fluorescence microscopy were performed. Classical MTT cell-viability assay79 was used to establish cytotoxicity of polymers P1, P5T and oligomer 5T toward two different kinds of cells, namely normal NIH-3T3 fibroblasts and lung cancer cells A549.The experimental cytotoxicity data were extrapolated using the exponential eqn (1):
 | (1) |
Fig. 7A summarizes the cell viability obtained after their incubation with P1, P5T and 5T at the concentration ranging from 0.01 to 1.0 mg mL−1. While 5T and P5T show non-toxic effect to NIH-3T3 cells up to 0.75 mg mL−1, P1 shows a cytotoxic effect starting with 0.5 mg mL−1 concentration. No cytotoxic activity against A549 cancerous cells was recorded for all the samples at an amount up to 0.5 mg mL−1. For the higher amount (0.75 mg mL−1), while 5T shows no effect, rather low viability was obtained for P5T and P1.The cytotoxic effects of these polymers could be associated with their structural peculiarities.
 | ||
| Fig. 7 (A) In vitro cell viability of NIH-3T3 cells (left side) and of A549 cells (right side) treated with various concentrations of 5T, P5T and P1; green dot line represents point of significant cytotoxicity (70% viability); (B) fluorescence microscope images of NIH-3T3 cells (left side) and A549 cells (right side), treated with 0.5 mg mL−1 of (a) 5T; (b) P5T; (c) P1 probes for 2 h. | ||
Thus, 5T and P5T contain less grafted PEG side chains on their backbone compared to P1 and, even if to date PEG has been considered a relatively benign, biocompatible material widely used in biomedical applications, however the effect of PEG incorporation in bioactive polymeric materials can be significant and complex, especially when the effect on cells fate is considered.80
If in the case of the nanoparticles the PEG chains grafting density is a critical factor modulating nanoparticles circulation time and non-specific cellular uptake,81 the concentration and molecular weight of PEG can also influence multiple properties including oxidative activity.82
The diverse PEG effects indicate that when the cells interact with PEG-containing biomaterials, the intracellular mechanism may change significantly as the amount of PEG varies.80 Recent studies evidenced a potential role of PEG as an intracellular exogenous provider of oxygen reactive species (ROS) which induce the increase of cells oxidative damage due to proteins carbonylation in an exponential manner especially when the PEG-containing materials are stored at physiological temperature and exposed to light.82 As in the case of our investigated materials the highest amount of PEG at the same molar concentration is in P1, this fact could explain its highest cytotoxicity toward NIH-3T3 cells. In contrast to the normal cells, cancer cells have a higher basal ROS output83 and the higher viability of A549 cancerous cells in the presence of P1 relative to NIH-3T3 cells could be explained based on this assumption. On the basis of the lower cytotoxicity and fluorescence properties of the synthesized polymers further investigation on their possible cell imaging applications were performed. After an incubation period of 2 h, of the polymers solutions (0.5 mg mL−1) with the cells, the fluorescence imaging studies were performed and the obtained images are presented in Fig. 7B.
The fluorescence images of the cells indicate that 5T and P5T can be efficiently internalized and dispersed in the cytoplasm by both type of cells, the normal NIH-3T3 fibroblasts (Fig. 7B, left side) and the lung cancer cells A549 (Fig. 7B, right side). If the fluorescence images obtained after A549 cells incubation with 0.5 mg mL−1 P1 suggest its labelling capability, this polymer cannot be used efficiently in the case of NIH-3T3 due to its increase cytotoxicity effect on fibroblast cells.
It is noticeable that 5T shows similar level of cytotoxic activity against cancerous and normal cell line at all the investigated concentration while at 0.75 mg mL−1 P5T showed a slightly specificity toward cancerous cells. These results shows that further optimization on their structure is needed to increase selective cytotoxicity to cancer cells over normal cells. Such goals can be obtained for macromonomers M1 or 5T by their copolymerization with an appropriate carboxyl-containing co-monomer for cancer cells targeting as previously reported,27e or by combination of 5T during water dissolution and nanoparticles formation with small amounts of other amphiphilic usual copolymer containing appropriate functional groups enabling for specific cells targeting.10
Conclusions
In this study we demonstrated the possibility to obtain fluorescent micellar nanoparticles readily dispersible in water based on oligo/polythiophene as photoactive material and PEG as water soluble one, using versatile thiophene chemistry. In terms of the materials design, the “grafting through” approach proved its suitability and flexibility for the synthesis of amphiphilic polymers in “hairy-rods” architecture, polymers able to mimic the “bottom-up” fabrication process found in nature, for an enhanced colloidal stability of their micellar nanoparticles as supramolecular structures.The methods used for polymerization or coupling of the synthesized PEG-containing macromonomers facilitated the obtainment of the polythiophenes with different length of the main chain concomitantly with various PEG side chains density. The investigations performed to characterize and to evaluate the oligo/polymers properties evidenced that the PEG side chains density is the crucial parameter influencing all the materials properties. Thus, a higher density of the PEG side chains induced a higher thermal stability of the reported materials in terms of their IDTs, and a higher size of the nanoparticles in water. Meanwhile we demonstrated that, by varying the value of this parameter was possible to tune the materials photophysical properties, a higher value for the λmax em being found for the polymer P1 by comparing with polymer P5T. The density of the PEG side chains also influences the investigated materials cytotoxicity, a lower viability for the investigated cell lines being registered for the polymer P1. The synthesized materials are remarkable from self-assembling property point of view, this capability-both in melt and in solution- being proved for polymer P1. Also it was demonstrated that the connecting way of the PEG side chains at the 3 position of the thiophene ring it is also very important. Thus, the introduction of a CH2 moiety between the ester functional group and the thiophene ring conducted to totally different photophysical properties of P2 by comparing with P1, its suitability for bio applications being excluded. The comparison of obtained nanoparticles photophysical properties with those of the unsubstituted polythiophene in thin film and with those of PThN in aqueous emulsion was a useful way to demonstrate that the PEG shell provides an electronic shielding from the external environment, thus preserving the core fluorescence.
From the application perspective, the values of DH, the high photostability, the absorbance and emission spectra of polythiophenes nanoparticles highlight their suitability for biological imaging, the broad absorption spectra providing flexibility in the choice of excitation wavelength. The most suitable for such applications seems to be the quinquethiophene 5T, that showed the highest values for Stokes shift, for molar absorption coefficient and especially the highest fluorescence quantum yield completed by a reasonable cytotoxicity and a slight specificity toward cancer cells. The present attempt offers an alternative to conducting polymer nanoparticles encapsulation in biocompatible matrix by nanoprecipitation. Moreover, the significant photophysical properties and the processing possibility of these fluorescent nanoparticles from eco-friendly solvents recommend them also for optoelectronic applications.
Acknowledgements
This paper is dedicated to the 65th anniversary of “Petru Poni” Institute of Macromolecular Chemistry of Romanian Academy, Iasi, Romania. One of the authors (A. D. B.) acknowledges the financial support given by European Social Fund “Cristofor I. Simionescu” Postdoctoral Fellowship Programme (ID POSDRU/89/1.5/S/55216), Sectorial Operational Programme Human Resources Development 2007–2013. We are grateful to Dr Luminita Marin from “Petru Poni” Institute of Macromolecular Chemistry, for her kindness regarding polarized optical microscopy investigations and for fruitful discussion. The authors are also grateful to Dr Mariana Pinteala, the head of Centre of Advanced Research in Bionanoconjugates and Biopolymers, at ”Petru Poni” Institute of Macromolecular Chemistry, for allowing access to some equipment for materials characterization. We also gratefully acknowledge to R. Bongartz at the Institute for Technical Chemistry at the Leibniz University of Hannover, Germany for her support in cells microscopy studies. The authors thank to Dr Demet Göen-Çolak from Istanbul Technical University-Turkey for critically reading the manuscript.References
- (a) S. Chen, S.-X. Cheng and R.-X. Zhuo, Macromol. Biosci., 2011, 11, 576–589 CrossRef CAS PubMed; (b) Z. Ahmad, A. Shah, M. Siddiq and H.-B. Kraatz, RSC Adv., 2014, 4, 17028–17038 RSC.
- H. Li, X. Zhang, X. Zhang, B. Yang, Y. Yang, Z. Huang and Y. Wei, RSC Adv., 2014, 4, 21588–21592 RSC.
- D. Gyawali, S. Zhou, R. T. Tran, Y. Zhang, C. Liu, L. Bai and J. Yang, Adv. Healthcare Mater., 2014, 3, 182–186 CrossRef CAS PubMed.
- B. Guo, L. Glavas and A.-C. Albertsson, Prog. Polym. Sci., 2013, 38, 1263–1286 CrossRef CAS PubMed.
- (a) T. V. Chirila, P. E. Rakoczy, K. L. Garrett, X. Lou and I. J. Constable, Biomaterials, 2002, 23, 321–342 CrossRef CAS; (b) R. Ravichandran, S. Sundarrajan, J. R. Venugopa, S. Mukherjee and S. Ramakrishna, J. R. Soc., Interface, 2010, 7, S559–S579 CrossRef CAS PubMed.
- A.-D. Bendrea, L. Cianga and I. Cianga, J. Biomater. Appl., 2011, 26, 3–85 CrossRef CAS PubMed.
- C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, Chem. Rev., 2012, 112, 4687–4735 CrossRef CAS PubMed.
- (a) D. Tuncel and H. V. Demir, Nanoscale, 2010, 2, 484–494 RSC; (b) J. Pecher and S. Mecking, Chem. Rev., 2010, 110, 6260–6279 CrossRef CAS PubMed; (c) A. Kaeser and A. P. H. J. Schenning, Adv. Mater., 2010, 22, 2985–2997 CrossRef CAS PubMed.
- E.-J. Park, T. Erdem, V. Ibrahimova, S. Nizamoglu, H. V. Demir and D. Tuncel, ACS Nano, 2011, 5, 2483–2492 CrossRef CAS PubMed.
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 2–26 Search PubMed.
- (a) V. Ibrahimova, S. Ekiz, O. Gezici and D. Tuncel, Polym. Chem., 2011, 2, 2818–2824 RSC; (b) O. Gezici, I. Durmaz, E. Bilget Guven, O. Unal, A. Ozgun, R. Cetin-Atalay and D. Tuncel, RSC Adv., 2014, 4, 1302–1309 RSC.
- F. Schütze, B. Stempfle, C. Jüngst, D. Wöll, A. Zumbusch and S. Mecking, Chem. Commun., 2012, 48, 2104–2106 RSC.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- S.-J. Park, S.-G. Kang, M. Fryd, J. G. Saven and S.-J. Park, J. Am. Chem. Soc., 2010, 132, 9931–9933 CrossRef CAS PubMed.
- H.-J. Kim, T. Kim and M. Lee, Acc. Chem. Res., 2011, 44, 72–82 CrossRef CAS PubMed.
- S.-H. Chou, H.-K. Tsao and Y.-J. Sheng, J. Chem. Phys., 2011, 134, 034904 CrossRef PubMed.
- P. Adriani, Y. Wang and W. L. Mattice, J. Chem. Phys., 1994, 100, 7718–7721 CrossRef CAS PubMed.
- H. Chen and E. Rukenstein, Soft Matter, 2012, 8, 1327–1333 RSC.
- S. C. Owen, D. P. Y. Chan and M. S. Shoichet, Nano Today, 2012, 7, 53–65 CrossRef CAS PubMed.
- (a) M. Zhang and A. H. E. Muller, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3461–3481 CrossRef CAS; (b) S. S. Sheiko, B. S. Sumerlin and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 759–785 CrossRef CAS PubMed.
- (a) J. A. Johnson, Y. Y. Lu, A. O. Burts, Y. H. Lim, M. G. Finn, J. T. Koberstein, N. J. Turro, D. A. Tirrell and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 559–566 CrossRef CAS PubMed; (b) Y.-L. Luo, L.-L. Zhang and F. Xu, Chem. Eng. J., 2012, 189, 431–442 CrossRef PubMed.
- P. Xu, H. Tang, S. Li, J. Ren, E. Van Kirk, W. J. Murdoch, M. Radosz and Y. Shen, Biomacromolecules, 2004, 5, 1736–1744 CrossRef CAS PubMed.
- G. Wegner, Macromol. Chem. Phys., 2003, 204, 347–357 CrossRef CAS.
- (a) U. Lauter, W. H. Meyer and G. Wegner, Macromolecules, 1997, 30, 2092–2101 CrossRef CAS; (b) P. Baum, W. H. Meyer and G. Wegner, Polymer, 2000, 41, 965–973 CrossRef CAS.
- I. Cianga and Y. Yagci, Prog. Polym. Sci., 2004, 29, 387–399 CrossRef CAS PubMed.
- (a) K. Ito, Prog. Polym. Sci., 1998, 23, 581–620 CrossRef CAS; (b) N. Hadjichristidis, M. Pitsikalis, H. Iatrou and S. Pispas, Macromol. Rapid Commun., 2003, 24, 979–1013 CrossRef CAS.
- (a) I. Cianga and Y. Yagci, Polym. Bull., 2001, 47, 17–24 CrossRef CAS; (b) I. Cianga, Y. Hepuzer and Y. Yagci, Polymer, 2002, 43, 2141–2149 CrossRef CAS; (c) A. L. Demirel, S. Yurteri, I. Cianga and Y. Yagci, Macromolecules, 2005, 38, 6402–6410 CrossRef CAS; (d) T. Uyar, I. Cianga, L. Cianga, F. Besenbacher and Y. Yagci, Mater. Lett., 2009, 63, 1638–1641 CrossRef CAS PubMed; (e) D. G. Colak, I. Cianga, D. O. Demirkol, O. Kozgus, E. I. Medine, S. Sakarya, P. Unak, S. Timur and Y. Yagci, J. Mater. Chem., 2012, 22, 9293–9300 RSC; (f) M. Yuksel, D. G. Colak, M. Akin, I. Cianga, M. Kukut, E. Ilker Medine, M. Can, S. Sakarya, P. Unak, S. Timur and Y. Yagci, Biomacromolecules, 2012, 13, 2680–2691 CrossRef CAS PubMed.
- (a) M. Wang, S. Zou, G. Guerin, L. Shen, K. Deng, M. Jones, G. C. Walker, G. D. Scholes and M. A. Winnik, Macromolecules, 2008, 41, 6993–7002 CrossRef CAS; (b) S. Das, D. P Chatterjee, S. Samanta and A. K. Nandi, RSC Adv., 2013, 3, 17540–17550 RSC; (c) J. Choi, C. R. Ruiz and E. E. Nesterov, Macromolecules, 2010, 43, 1964–1974 CrossRef CAS; (d) S. Das, S. Samanta, D. P. Chatterjee and A. K. Nandi, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1417–1427 CrossRef CAS.
- Handbook of Thiophene-based Materials: Applications in Organic Electronics and Photonics, ed. I. F. Perepichka and D. F. Perepichka, John Wiley and Sons, 2009 Search PubMed.
- A. Å. Sigurdson, C. J. Klingstedt, T. Grathwohl, S. Bolmont, T. Dickstein, D. L. Glimsdal, E. Prokop, S. Lindgren, M. Konradsson, P. Holtzman, D. M. Hof, P. R. Heppner, F. L. Gandy, S. Jucker, M. Aguzzi, A. Hammarstrom and P. Nilsson, ACS Chem. Biol., 2009, 4, 673–684 CrossRef PubMed.
- (a) M. Zambianchi, F. Di Maria, A. Cazzato, G. Gigli, M. Piacenza, F. Della Sala and G. Barbarella, J. Am. Chem. Soc., 2009, 131, 10892–10900 CrossRef CAS PubMed; (b) G. Yang, L. Liu, Q. Yang, F. Lv and S. Wang, Adv. Funct. Mater., 2012, 22, 736–743 CrossRef CAS.
- C. T. Chang, et al., US Pat., no. 5,578,636/26 November 1996.
- C. Xing, G. Yang, L. Liu, Q. Yang, F. Lv and S. Wang, Small, 2012, 8, 525–529 CrossRef CAS PubMed.
- C. Xing, L. Liu, H. Tang, X. Feng, Q. Yang, S. Wang and G. C. Bazan, Adv. Funct. Mater., 2011, 21, 4058–4067 CrossRef CAS.
- (a) D. F. Li, H. J. Wang, J. X. Fu, W. Wang, X. S. Jia and J. Y. Wang, J. Phys. Chem. B, 2008, 112, 16290–16299 CrossRef CAS PubMed; (b) M. Waugaman, B. Sannigrahi, P. McGeady and I. M. Khan, Eur. Polym. J., 2003, 39, 1405–1412 CrossRef CAS; (c) M. M. Perez-Madrigal, E. Armelin, L. J. del Valle, F. Estrany and C. Aleman, Polym. Chem., 2012, 3, 979–991 RSC; (d) A.-D. Bendrea, G. Fabregat, J. Torras, S. Maione, L. Cianga, L. J. del Valle, I. Cianga and C. Aleman, J. Mater. Chem. B, 2013, 1, 4135–4145 RSC.
- (a) C. I. Simionescu, I. Cianga, M. Ivanoiu, A. Duca, I. Cocarla and M. Grigoras, Eur. Polym. J., 1999, 35, 587–599 CrossRef CAS; (b) L. Cianga and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 995–1004 CrossRef CAS; (c) F. Yilmaz, L. Cianga, Y. Guner, L. Toppare and Y. Yagci, Polymer, 2004, 45, 5765–5774 CrossRef CAS PubMed.
- (a) F. Yilmaz, I. Cianga, K. Ito, T. Senyo and Y. Yagci, Macromol. Rapid Commun., 2003, 24, 316–319 CrossRef CAS; (b) L. Cianga, A. Sarac, K. Ito and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 479–492 CrossRef CAS.
- (a) P. Howes, M. Green, J. Levitt, K. Suhling and M. Hughes, J. Am. Chem. Soc., 2010, 132, 3989–3996 CrossRef CAS PubMed; (b) P. K. Kandel, L. P. Fernando, C. Ackroyd and K. A. Christensen, Nanoscale, 2011, 3, 1037–1045 RSC; (c) F. Su, R. Alam, Q. Mei, Y. Tian and D. R. Meldrum, PLoS One, 2011, 6, e24425 CAS; (d) Y. Li, J. Liu, B. Liu and N. Tomczak, Nanoscale, 2012, 4, 5694–5702 RSC.
- (a) P. V. Torchilin, J. Controlled Release, 2001, 73, 137 CrossRef; (b) M. Siauw, B. S. Hawkett and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 187–198 CrossRef CAS.
- (a) J. H. Yao, K. Y. Mya, L. Shen, B. P. He, L. Li, Z. H. Li, Z.-K. Chen, X. Li and K. P. Loh, Macromolecules, 2008, 41, 1438–1443 CrossRef CAS; (b) S.-C. Liao, C.-S. Lai, D.-D. Yeh, M. H. Rahman, C.-S. Hsu, H.-L. Chen and S.-A. Chen, React. Funct. Polym., 2009, 69, 498–506 CrossRef CAS PubMed; (c) K.-Y. Pu, K. Li and B. Liu, Adv. Funct. Mater., 2010, 20, 2770–2777 CrossRef CAS; (d) A.-D. Bendrea, L. Cianga, E. G. Hitruc, I. Titorencu and I. Cianga, Mater. Plast., 2013, 50, 71–78 CAS.
- (a) J. M. Lee, S. J. Lee, Y. J. Jung and J. H. Kim, Curr. Appl. Phys., 2008, 8, 659–663 CrossRef PubMed; (b) S. J. Lee, J. M. Lee, I. W. Cheong, H. S. Lee and J. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2097–2107 CrossRef CAS; (c) S. J. Lee, J. M. Lee, H.-Z. Cho, W. G. Koh, I. W. Cheong and J. H. Kim, Macromolecules, 2010, 43, 2484–2489 CrossRef CAS.
- D. Zehm, A. Laschewsky, P. Heunemann, M. Gradzielski, S. Prevost, H. Liang, J. P. Rabe and J. F. Lutz, Polym. Chem., 2011, 2, 137–147 RSC.
- (a) A.-D. Bendrea, L. Cianga, E. G. Hitruc and I. Cianga, Int. J. Polym. Anal. Charact., 2013, 18, 189–198 CrossRef CAS; (b) A.-D. Bendrea, G. Fabregat, L. Cianga, F. Estrany, L. J. del Valle, I. Cianga and C. Aleman, Polym. Chem., 2013, 4, 2709–2723 RSC; (c) A.-D. Bendrea, L. Cianga and I. Cianga, Rev. Roum. Chim., 2013, 58, 153–160 CAS.
- (a) O. Papila, L. Toppare, Y. Yagci and L. Cianga, Int. J. Polym. Anal. Charact., 2004, 9, 13–28 CrossRef CAS; (b) S. Alkan, L. Toppare, Y. Hepuzer and Y. Yagci, J. Polym. Sci., Polym. Chem. Ed., 1999, 37, 4218–4225 CrossRef CAS; (c) I. Kerman, L. Toppare, F. Yilmaz and Y. Yagci, J. Macromol. Sci., Part A: Pure Appl. Chem., 2005, 42, 509–520 CrossRef PubMed; (d) A. C. Kucuk, F. Yilmaz, H. Can, H. Durmaz, A. Kosemen and A. E. Muftuoglu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4180–4192 CAS; (e) I. Levesque and M. Leclerc, Chem. Mater., 1996, 8, 2843–2849 CrossRef CAS; (f) H. B. Yildiz, S. Kiralp, L. Toppare, Y. Yagci and K. Ito, Mater. Chem. Phys., 2006, 100, 124–127 CrossRef CAS PubMed.
- H. B. Liu, A. Jiang, J. A. Guo and K. E. Uhrich, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 703–711 CrossRef CAS.
- S. M. Paterson, D. H. Brown, T. V. Chirila, I. Keen, A. K. Whittaker and M. V. Baker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4084–4092 CrossRef CAS.
- T. Yamamoto, Bull. Chem. Soc. Jpn., 2010, 83, 431–455 CrossRef CAS.
- (a) H. Masuda and K. Kaeriyama, Makromol. Chem., Rapid Commun., 1992, 13, 461–465 CrossRef CAS; (b) T. Sakaguchi, S. Tominaga and T. Hashimoto, Polymer, 2011, 52, 2163–2169 CrossRef CAS PubMed.
- (a) H. Mao and S. Holdcroft, Macromolecules, 1992, 25, 554–558 CrossRef CAS; (b) T. Yamamoto, A. Morita, Y. Miyazaki, T. Maruyama, H. Wakayama, Z.-H. Zhou, Y. Nakamura, T. Kanbara, S. Sasaki and K. Kubota, Macromolecules, 1992, 25, 1214–1223 CrossRef CAS; (c) H. Mao, B. Xu and S. Holdcroft, Macromolecules, 1993, 26, 1163–1164 CrossRef CAS; (d) G. Barbarella, A. Bongini and M. Zambianchi, Macromolecules, 1994, 27, 3039–3045 CrossRef CAS; (e) T. A. Chen, X. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233–244 CrossRef CAS.
- R. S. Becker, J. S. de Melo, A. L. Macüanita and F. Elisei, J. Phys. Chem., 1996, 100, 18683–18695 CrossRef CAS.
- (a) T. Yamamoto, K. Sanechika and A. Yamamoto, Bull. Chem. Soc. Jpn., 1983, 56, 1497–1502 CrossRef CAS; (b) Z. Mo, K.-B. Lee, Y. B. Moon, M. Kobayashi, A. J. Heeger and F. Wudl, Macromolecules, 1985, 18, 1972–1977 CrossRef CAS.
- E. Armelin, O. Bertran, F. Estrany, R. Salvatella and C. Aleman, Eur. Polym. J., 2009, 45, 2211–2221 CrossRef CAS PubMed.
- M. Bjerring, J. S. Nielsen, N. C. Nielsen and F. C. Krebs, Macromolecules, 2007, 40, 6012–6013 CrossRef CAS.
- (a) W. Porzio, S. Destri, M. Mascherpa and S. Bruckner, Acta Polym., 1993, 44, 266–272 CrossRef CAS; (b) A. J. Lovinger, D. D. Davis, A. Dodabalapur and H. E. Katz, Chem. Mater., 1996, 8, 2836–2838 CrossRef CAS; (c) M. Melucci, M. Gazzano, G. Barbarella, M. Cavallini, F. Biscarini, P. Maccagnani and P. Ostoj, J. Am. Chem. Soc., 2003, 125, 10266–10274 CrossRef CAS PubMed.
- C. He, J. Sun, T. Zhao, Z. Hong, X. Zhuang, X. Chen and X. Jing, Biomacromolecules, 2006, 7, 252–258 CrossRef CAS PubMed.
- E. Arenda, A. Leiva, L. Gargallo, N. Hadjichristidis, I. Mondragon and D. Radic, Polym. Eng. Sci., 2012, 52, 1128–1136 Search PubMed.
- K. Knop, G. M. Pavlov, R. Tobias, K. Martin, D. Pretzel, B. O. Jahn, D. H. Scharf, A. A. Brakhage, V. Makarov, U. Mollmann, F. H. Schachera and U. S. Schubert, Soft Matter, 2013, 9, 715–726 RSC.
- T. Rager, W. H. Meyer, G. Wegner and M. A. Winnik, Macromolecules, 1997, 30, 4911 CrossRef CAS.
- A. P.-Z. Clark, C. Shi, B. C. Ng, J. N. Wilking, A. L. Ayzner, A. Z. Stieg, B. J. Schwartz, T. G. Mason, Y. Rubin and S. H. Tolbert, ACS Nano, 2013, 7, 962–977 CrossRef CAS PubMed.
- C. R. Heald, S. Stolnik, K. S. Kujawinski, C. De Matteis, M. C. Garnett, L. Illum, S. S. Davis, S. C. Purkiss, R. J. Barlow and P. R. Gellert, Langmuir, 2002, 18, 3669–3675 CrossRef CAS.
- Z. Yang, Y. Yuan, R. Jiang, N. Fu, X. Lu, C. Tian, W. Hu, Q. Fan and W. Huang, Polym. Chem., 2014, 5, 1372–1380 RSC.
- M. G. Mohamed, C.-C. Cheng, Y.-C. Lin, C.-W. Huang, F.-H. Lu, F.-C. Chang and S.-W. Kuo, RSC Adv., 2014, 4, 21830–21839 RSC.
- O. Tirosh, Y. Barenholz, J. Katzhendler and A. Priev, Biophys. J., 1998, 74, 1371–1379 CrossRef CAS.
- J. B. Matson and R. H. Grubbs, J. Am. Chem. Soc., 2008, 130, 6731–6733 CrossRef CAS PubMed.
- W. Abdelwahed, G. Degobert, S. Stainmesse and H. Fessi, Adv. Drug Delivery Rev., 2006, 58, 1688–1713 CrossRef CAS PubMed.
- K. Lee, H.-J. Kim and J. Kim, Adv. Funct. Mater., 2012, 22, 1076–1086 CrossRef CAS.
- S. Han, M. Hagiwara and T. Ishizone, Macromolecules, 2003, 36, 8312–8319 CrossRef CAS.
- (a) M. S. Nikolic, C. Olsson, A. Salcher, A. Kornowski, A. Rank, R. Schubert, A. Frömsdorf, H. Weller and S. Förster, Angew. Chem., Int. Ed., 2009, 48, 2752–2754 CrossRef CAS PubMed; (b) K. Zhang, M. Jiang and D. Chen, Prog. Polym. Sci., 2012, 37, 445–486 CrossRef CAS PubMed.
- E. Jäger, A. Jäger, T. Etrych, F. C. Giacomelli, P. Chytil, A. Jigounov, J. L. Putaux, B. Říhová, K. Ulbrich and P. Štěpánek, Soft Matter, 2012, 8, 9563–9575 RSC.
- (a) Y. Wang, W. B. Euler and B. L. Lucht, Chem. Commun., 2004, 686–687 RSC; (b) R. van Beek, L. W. Jenneskens, A. N. Zdravkova, J. P. J. M. van der Eerden and C. A. van Walree, Macromol. Chem. Phys., 2005, 206, 1006–1014 CrossRef CAS.
- I. F. Perepichka, D. F. Perepichka, H. Meng and F. Wudl, Adv. Mater., 2005, 17, 2281–2305 CrossRef CAS.
- (a) F. Brustolin, F. Goldoni, E. W. Meijer and N. A. J. M. Sommerdijk, Macromolecules, 2002, 35, 1054–1059 CrossRef CAS; (b) J. R. Matthews, F. Goldoni, A. P. H. J. Schenning and E. W. Meijer, Chem. Commun., 2005, 5503–5505 RSC.
- (a) Y. Li, A. Nese, X. Hu, N. V. Lebedeva, T. W. LaJoie, J. Burdyńska, M. C. Stefan, W. You, W. Yang, K. Matyjaszewski and S. S. Sheiko, ACS Macro Lett., 2014, 3, 738–742 CrossRef CAS; (b) G. Nagarjuna, M. Baghgar, J. A. Labastide, D. D. Algaier, M. D. Barnes and D. Venkataraman, ACS Nano, 2012, 6, 10750–10758 CAS.
- N. Kurokawa, H. Yoshikawa, N. Hirota, K. Hyodo and H. Masuhara, ChemPhysChem, 2004, 5, 1609–1615 CrossRef CAS PubMed.
- S. Natarajan and S. H. Kim, Thin Solid Films, 2006, 496, 606–611 CrossRef CAS PubMed.
- G. Sun, M. Y. Berezin, J. Fan, H. Lee, J. Ma, K. Zhang, K. L. Wooley and S. Achilefu, Nanoscale, 2010, 2, 548–558 RSC.
- Z. Hashim, P. Howes and M. Green, J. Mater. Chem., 2011, 21, 1797–1803 RSC.
- P. Howes and M. Green, Photochem. Photobiol. Sci., 2010, 9, 1159–1166 CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- H.-J. Sung, A. Luk, N. S. Murthy, E. Liu, M. Jois, A. Joy, J. Bushman, P. V. Mogheb and J. Kohn, Soft Matter, 2010, 6, 5196–5205 RSC.
- S. Hak, E. Helgesen, H. H. Hektoen, E. M. Huuse, P. A. Jarzyna, W. J. M Mulder, O. Haraldseth and C. de Lange Davies, ACS Nano, 2012, 6, 5648–5658 CrossRef CAS PubMed.
- H. J. Sung, P. Chandra, M. D. Treiser, E. Liu, C. P. Iovine, P. V. Moghe and J. Kohn, J. Cell. Physiol., 2009, 218, 549–557 CrossRef CAS PubMed.
- D. Trachootham, Y. Zhou, H. Zhang, Y. Demizu, Z. Chen, H. Pelicano, P. J. Chiao, G. Achanta, R. B. Arlinghaus, J. Liu and P. Huang, Cancer Cell, 2006, 10, 241–252 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details concerning the intermediates and macromonomers synthesis, supplementary discussion concerning to the IR characterization of the reported compounds, supporting data and discussion for GPC measurements and HLB calculations, two supplementary table, ten supplementary figures and supplementary references. See DOI: 10.1039/c4ra10734a |
| ‡ Both authors contributed equally as first authors to this work. |
| This journal is © The Royal Society of Chemistry 2014 |