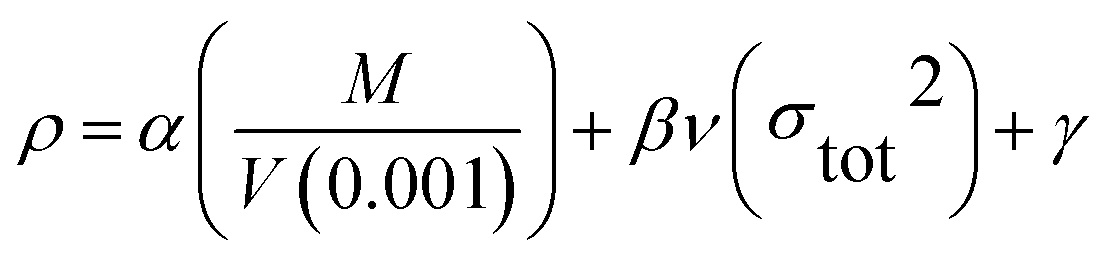DOI:
10.1039/C4RA10548F
(Paper)
RSC Adv., 2014,
4, 53000-53009
Designing and screening novel explosives with high energy and low sensitivity by appropriately introducing N-oxides, amino groups, and nitro groups into s-heptazine
Received
16th September 2014
, Accepted 3rd October 2014
First published on 3rd October 2014
Abstract
We presented a useful strategy to design novel explosives by incorporating N-oxides, nitro groups, and amino groups into s-heptazine. Five new high explosives s-heptazine-1,3,4,6,7,9-hexaoxides (HTO), trinitroheptazine-1,3,4,6,7,9-hexaoxides (TNHTO), aminodinitroheptazine-1,3,4,6,7,9-hexaoxides (ADNHTO), diaminonitroheptazine-1,3,4,6,7,9-hexaoxides (DANHTO), and triaminoheptazine-1,3,4,6,7,9-hexaoxides (TAHTO) were designed. Their energetic properties and sensitivity were estimated by using density functional theory and compared with some famous explosives like CL-20, ONC, HMX, and TNT. All five designed new explosives have much higher detonation performance than s-heptazine and HMX, indicating that symmetrically introducing six N-oxides into s-heptazine is a very effective strategy to improve the explosive performance. DANHTO and TAHTO have comparable detonation performance with CL-20 or ONC and TAHTO has lower sensitivity than TNT, indicating that appropriately incorporating N-oxides, amino groups, and nitro groups into s-heptazine can generate new explosives with excellent performance and low sensitivity. This strategy may be used to design and develop other new energetic materials.
1 Introduction
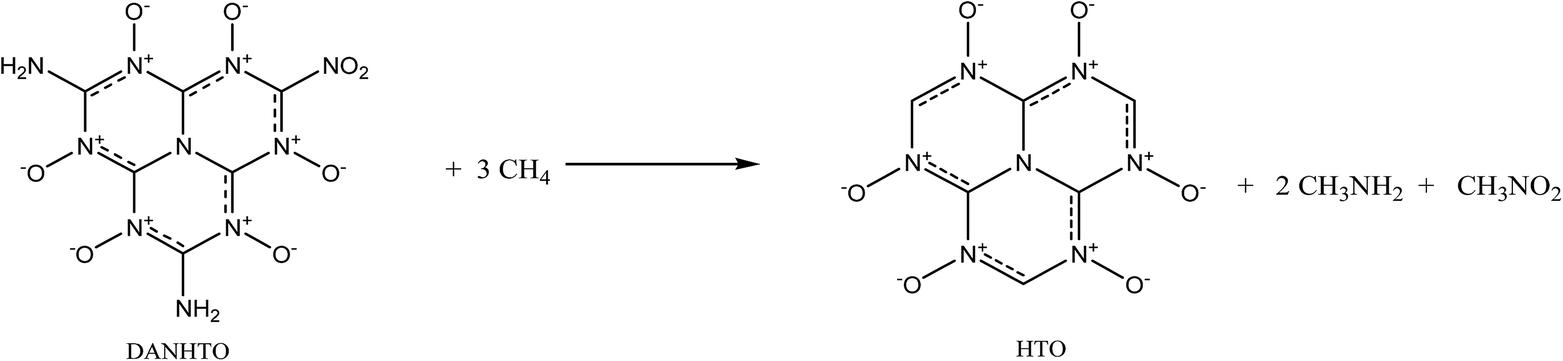
1,3,4,6,7,9-hexaazacycl[3,3,3]azine1–3 (s-heptazine or tri-s-triazine, C6H3N7, Fig. 1) is a conjugated symmetrical planar molecule with high nitrogen content (56.7%), high heat of formation (HOF), and high thermal stability, making it a valuable precursor used to design and synthesize new high explosives with good detonation properties and low sensitivity. Some of its derivatives like 2,5,8-trichloro-s-heptazine,4 2,5,8-triamino-s-heptazine,5 and 2,5,8-triazido-s-heptazine6 have been synthesized successfully. Although many of them have outstanding thermal stability, their explosive performances are not high and obviously lower than those of two famous and widely used explosives 1,3,5-trinitro-1,3,5-triazinane (RDX) and 1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX).7 This is because most of the s-heptazine derivatives have low oxygen balance and densities, which are two very important factors related closely to the detonation properties of the explosives. In general, the lower the oxygen balance and density are, the smaller the detonation velocity and pressure are, and the poorer the performance of the explosive is. Thus, it is necessary to figure out some useful strategies to improve the energetic properties of s-heptazine derivatives by increasing the oxygen balance and density. One effective way is to introduce N-oxides into s-heptazine. This method has been successfully applied into two azacylco compounds (pyrazine8 and pyridine9) which have much lower nitrogen content than s-heptazine. Based on the above discussion, a new compound 1,3,4,6,7,9-hexaazacycl[3,3,3]azine-1,3,4,6,7,9-hexaoxides (or s-heptazine-1,3,4,6,7,9-hexaoxides, HTO) is designed and its oxygen balance (−45%) is much higher than that of s-heptazine (−125%). Its structure, density, detonation performance, and other properties should be investigated further.
 |
| | Fig. 1 Molecular frameworks of s-heptazine, HTO, TNHTO, ADNHTO, DANHTO, and TAHTO. | |
In the past several decades, theoretical studies based on quantum chemical treatment have gained acceptance as a useful research tool to screen the candidates of insensitive high explosives, thereby avoiding a lot of expensive and dangerous experimental tests. They can also provide the relationships between molecular structure and property, which in turn can help design better and more efficient laboratory tests. Accordingly, theoretical design of candidate compounds with high energy and insensitivity is the primary step for synthesizing new explosives.10
In this work, one novel explosive HTO was designed by symmetrically introducing six N-oxides into the s-heptazine. Then, the structure–property relationships were investigated by incorporating different numbers of nitro groups and amino groups into the HTO system. Finally, the electronic structure, heat of formation, spectral properties, density, energetic properties, pyrolysis mechanism, and sensitivity of HTO and its derivatives were studied by using density functional theory (DFT). Our main purpose is to look for novel explosives with high density, high detonation performance, good thermal stability, and low sensitivity by combining N-oxides, amino group, and/or nitro group in the s-heptazine.
2 Computational method
The DFT-B3LYP method with 6-311G(d,p) basis set was successfully used to predict the HOFs of many organic systems via isodesmic reactions.11–19 The isodesmic reactions used to obtain the heats of formation of all substituted-HTO derivatives at 298 K are as follows: | |

| (1) |
| |
| (2) |
| |
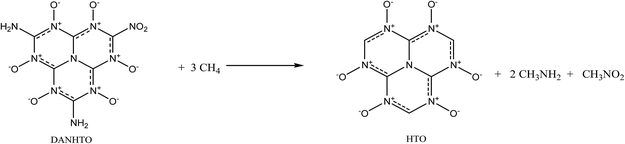
| (3) |
| |

| (4) |
For the isodesmic reaction, heat of reaction ΔH298 at 298 K could be calculated from the following equation:
| | |
ΔH298 = ΔHf,P − ΔHf,R
| (5) |
where Δ
Hf,R and Δ
Hf,P are the HOFs of reactants and products at 298 K, respectively. As the experimental HOFs of
s-heptazine and HTO are unavailable, additional calculations were carried out for the atomization reaction C
aH
bO
cN
d → aC(g) + bH(g)+ cO(g) + dN(g) by using the CBS-4M theory to get an accurate value of Δ
Hf. The experimental heats of formation of reference compounds CH
4, CH
3NO
2, and NH
3 are available. Now the most important task is to compute Δ
H298. The Δ
H298 can be calculated using the following expression:
| | |
ΔH298 = ΔE298 + Δ(PV) = ΔE0 + ΔEZPE + ΔET + ΔnRT
| (6) |
where Δ
E0 is the change in total energy between the products and reactants at 0 K; Δ
EZPE is the difference between the zero-point energies (ZPE) of the products and reactants at 0 K; Δ
ET is thermal correction from 0 to 298 K. The Δ(
PV) value in
eqn (6) is
PV work term and equals Δ
nRT for the reactions of ideal gas. For the isodesmic reaction in this work, Δ
n = 0, so Δ(
PV) = 0.
According to Hess's law of constant heat summation,20 the solid-phase heat of formation can be obtained from the gas-phase heat of formation (ΔHf,gas) and heat of sublimation (ΔHsub):
| | |
ΔHf,solid = ΔHf,gas − ΔHsub
| (7) |
Politzer et al.21,22 reported that the heat of sublimation correlates with the molecular surface area and the electrostatic interaction index for energetic compounds. The empirical expression of the approach is as follows:
| | |
ΔHsub = aA2 + b(νσtot2)0.5 + c
| (8) |
where
A is the surface area of the 0.001 electrons bohr
−3 isosurface of the electronic density of the molecule,
ν describes the degree of balance between positive potential and negative potential on the isosurface, and is a measure of the variability of the electrostatic potential on the molecular surface. The coefficients
a,
b, and
c have been determined by Rice
et al.:
a = 2.670 × 10
−4 kcal mol
−1 A
−4,
b = 1.650 kcal mol
−1, and
c = 2.966 kcal mol
−1.
23 The descriptors
A,
ν, and
σtot2 were calculated by using the computational procedures proposed by Bulat
et al.24 This approach has been demonstrated to predict reliably the heats of sublimation of many energetic organic compounds.
24,25
The detonation velocity and pressure were estimated by the Kamlet–Jacobs equations26 as
| |
D = 1.01(N![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) 1/2Q1/2)1/2(1 + 1.30ρ) 1/2Q1/2)1/2(1 + 1.30ρ)
| (9) |
| | |
P = 1.558ρ2N1/2Q1/2
| (10) |
where each term in the
eqn (8) and
(9) is defined as follows:
D, the detonation velocity (km s
−1);
P, the detonation pressure (GPa);
N, the moles of detonation gases per gram explosive; the average molecular weight of these gases;
Q, the heat of detonation (cal g
−1); and
ρ, the loaded density of explosives (g cm
−3). For known explosives, their
Q and
ρ can be measured experimentally; thus their
D and
P can be calculated according to
eqn (9) and
eqn (10). However, for some compounds, their
Q and
ρ cannot be evaluated from experimental measures. Therefore, to estimate their
D and
P, we first need to calculate their
Q and
ρ.
The theoretical density was obtained using an improved equation proposed by Politzer et al.27 in which the interaction index νσtot2 was introduced:
| |
 | (11) |
where
M is the molecular mass (g mol
−1) and
V(0.001) is the volume of the 0.001 electrons bohr
−3 contour of electronic density of the molecule (cm
3 molecule
−1). The coefficients
α,
β, and
γ are 0.9183, 0.0028, and 0.0443, respectively.
The strength of bonding, which could be evaluated by bond dissociation energy (BDE), is fundamental to understand chemical processes.28 The BDE plays an important role on judging the thermal stability of energetic materials. Generally, the higher energy for breaking a bond is, the stronger the bond is, the more difficult the bond becomes a trigger bond; that is to say, the corresponding compound is more stable, and its sensitivity is lower. Therefore, the calculated BDE could be used to likely measure the relative order of thermal stability for energetic compounds. The energy required for bond homolysis at 298 K and 1 atm corresponds to the enthalpy of reaction A–B (g) → A (g) + B (g), which is the bond dissociation enthalpy of the molecule A–B by definition.29 For many organic molecules, the terms “bond dissociation energy” and “bond dissociation enthalpy” usually appear interchangeably in the literature.30 Thus, at 0 K, the homolytic bond dissociation energy can be given in terms of eqn (12):
| | |
BDE0(A–B) = E0(A) + E0(B) − E0(A–B)
| (12) |
The bond dissociation energy with zero-point energy (ZPE) correction can be calculated by eqn (13):
| | |
BDE(A–B)ZPE = BDE0(A–B) + ΔEZPE
| (13) |
where Δ
EZPE is the difference between the ZPEs of the products and the reactants.
The sensitivity of explosives is a parameter which determines how easily a fast reaction can be initiated in a sample when subjected to an external stimulus. Among all the sensitivities, impact sensitivity is most commonly measured. The impact sensitivity is affected by many factors including its structure, physical state, crystalline form, and size and grain of crystal. In addition, different measurement conditions could strongly affect the sensitivity measurements. This is why the results of experimental impact sensitivity are not reliable and repeatable usually. If a series of energetic materials have similar structure or similar thermal decomposition mechanism, their intrinsic structure become main factor in determining their sensitivity and the effects of other factors can be counteracted. Therefore, theoretical methods can be used as a tool to predict the impact sensitivity of new designed energetic compounds with similar structure in the absence of experimental results. Lately, the impact sensitivity (h50, cm) can be estimated by eqn (14):31
| | |
h50 = α[Veff − V(0.002)]1/3 + βvσtot2 + γ
| (14) |
V(0.002) and V(0.003) are defined as the volume enclosed by the 0.002 and 0.003 electrons bohr−3 contour of the molecule's electronic density, respectively. ν describes the degree of balance between positive potential and negative potential on the isosurface. σtot2 is a measure of the variability of the electrostatic potential on the molecular surface. The coefficients α, β, and γ are −234.83, −3.197, and 962.0, respectively. Veff can be calculated exactly from the dimensions of the unit cell and the number of molecules that it encompasses, or alternatively by the formula:
where
M is the molecular mass and
ρ is the crystal density.
The calculations were performed at the B3LYP/6-311G(d,p) level with the Gaussian 03 package.32 In the geometry optimization, the maximum force was converged less than 0.00045 eV Å−1, the RMS force less than 0.0003 eV Å−1, the maximum displacement less than 0.0018 Å, and the RMS displacement less than 0.0012 Å. All the optimized structures were characterized to be true local energy minima on their potential energy surfaces without imaginary frequencies. The infrared (IR), and ultraviolet-visible (UV-VIS) spectrums were calculated at the B3LYP/6-311G(d,p) method.
3 Results and discussion
3.1 Introduction of six N-oxides into s-heptazine symmetrically
In this section, one novel explosive HTO (Fig. 1) was designed by symmetrically introducing six N-oxides into the s-heptazine. Then, its structure, heat of formation, density, detonation properties, thermal stability, and sensitivity are studied systematically. Table 1 lists the calculated bond lengths of s-heptazine and HTO. The computed bond lengths of s-heptazine are very close to the experimental values,1 indicating that our calculated results are reliable. The C–N bond lengths of HTO range from 1.32 to 1.43 Å, lying between the common C–N bond length (1.48 Å)33 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond length (1.28 Å).33 Moreover, the bond lengths of the N–O bonds linked to the ring are around 1.26 Å, which are also between the normal N–O bond length (1.40 Å)33 and NO bond length (1.21 Å).33 The optimized structure of HTO is a big planar molecule, as displayed in Fig. 2. Table 2 lists the calculated solid HOFs, densities, D, and P of s-heptazine, HTO, RDX,7,35 HMX,7,35 2,4,6-trinitrophenol (picric acid, PA),7 1-methyl-2,4,6-trinitrobenzene (TNT),7 and 2,6-diamino-3,5-dinitropyrazin-1-oxide (LLM-105).36–38 It is found that our calculated results on RDX, HMX, PA, TNT, and LLM-105 are very close to available experimental values.1,7,34–37 The HOF, density, D, and P of HTO are markedly higher than those of s-heptazine, suggesting that the introduction of six N-oxides symmetrically into s-heptazine is a very effectively strategy to improve its explosive performance.
N bond length (1.28 Å).33 Moreover, the bond lengths of the N–O bonds linked to the ring are around 1.26 Å, which are also between the normal N–O bond length (1.40 Å)33 and NO bond length (1.21 Å).33 The optimized structure of HTO is a big planar molecule, as displayed in Fig. 2. Table 2 lists the calculated solid HOFs, densities, D, and P of s-heptazine, HTO, RDX,7,35 HMX,7,35 2,4,6-trinitrophenol (picric acid, PA),7 1-methyl-2,4,6-trinitrobenzene (TNT),7 and 2,6-diamino-3,5-dinitropyrazin-1-oxide (LLM-105).36–38 It is found that our calculated results on RDX, HMX, PA, TNT, and LLM-105 are very close to available experimental values.1,7,34–37 The HOF, density, D, and P of HTO are markedly higher than those of s-heptazine, suggesting that the introduction of six N-oxides symmetrically into s-heptazine is a very effectively strategy to improve its explosive performance.
Table 1 Calculated bond lengths (Å) of s-heptazine, HTO, TNHTO, ADNHTO, DANHTO, and TAHTO
| Compound |
(C–N)ring |
C–H |
N–O linked to the ring |
N–O in NO2 |
C–NO2 |
C–NH2 |
N–H |
| Experimental values from ref. 1. |
| s-heptazine |
1.33–1.41 (1.30–1.39a) |
1.09 (1.01–1.12a) |
|
|
|
|
|
| HTO |
1.32–1.43 |
1.07 |
1.25–1.27 |
|
|
|
|
| TNHTO |
1.33–1.42 |
|
1.25–1.26 |
1.21 |
1.48 |
|
|
| ADNHTO |
1.35–1.39 |
|
1.27–1.28 |
1.21 |
1.47–1.48 |
1.31 |
1.01 |
| DANHTO |
1.36–1.39 |
|
1.27–1.29 |
1.21 |
1.47 |
1.31 |
1.01 |
| TAHTO |
1.37–1.41 |
|
1.30 |
|
|
1.32 |
1.01 |
 |
| | Fig. 2 (a) The optimized structures of HTO, TNHTO, ADNHTO, DANHTO, and TAHTO. (b) The perspective view of A HTO, TNHTO, ADNHTO, DANHTO, and TAHTO from another viewpoint. White, red, blue, and gray spheres stand for H, O, N, and C atoms, respectively. | |
Table 2 Solid-phase HOFs (kJ g−1), oxygen balance (OB, %), densities (ρ, g cm−3), D (km s−1), and P (GPa) of HTO, TNHTO, ADNHTO, DANHTO, TAHTO, RDX, and HMX
| Compound |
HOFs |
OB |
ρ |
D |
P |
| Experimental values from ref. 1. Experimental values from ref. 34. Experimental values from ref. 35. Experimental values from ref. 7. Experimental values from ref. 36. Experimental values from ref. 37. Computed values from ref. 38. |
| s-heptazine |
2.4 |
−125 |
1.67 (1.69a) |
5.6 |
13.3 |
| HTO |
4.9 |
−45 |
1.94 |
9.5 |
42.0 |
| TNHTO |
3.7 |
0 |
2.01 |
10.3 |
49.7 |
| ADNHTO |
3.5 |
−13 |
2.03 |
10.1 |
48.2 |
| DANHTO |
3.4 |
−28 |
2.04 |
9.8 |
45.4 |
| TAHTO |
3.3 |
−46 |
2.03 |
9.5 |
43.1 |
| RDX |
0.4 (0.4b) |
−22 |
1.81 (1.82c) |
8.8 (8.7c) |
34.7 (34.5c) |
| HMX |
0.4 (0.4b) |
−22 |
1.90 (1.90c) |
9.2 (9.1c) |
39.1 (39.0c) |
| PA |
−1.0 (−1.1d) |
−45 |
1.79 (1.77d) |
7.5 (7.4d) |
26.0 |
| TNT |
−0.3 (−0.2d) |
−74 |
1.66 (1.65d) |
7.0 (6.9d) |
21.1 |
| LLM-105 |
0.1 (0.1g) |
−37 |
1.89 (1.92e) |
8.5 (8.6f) |
31.2 |
Table 3 lists the calculated BDE of the relatively weak bonds of s-heptazine and HTO. The C–N has the lowest BDE among the calculated bonds. The BDE of s-heptazine is higher than that of HTO, suggesting that the former has better thermal stability than the later. Table 4 lists the h50 values of s-heptazine and HTO. It is seen in Table 4 that HTO has higher h50 value than s-heptazine, indicating that HTO has higher sensitivity than s-heptazine.
Table 3 Calculated BDE (kJ mol−1) of the relatively weak bonds of HTO, TNHTO, ADNHTO, DANHTO, TAHTO, RDX, and HMX
| Compound |
(C–N)ring |
C–NO2 |
C–NH2 |
N–NO2 |
| Calculated values from ref. 39. |
| s-heptazine |
466.7 |
|
|
|
| HTO |
400.1 |
|
|
|
| TNHTO |
288.5 |
109.1 |
|
|
| ADNHTO |
293.7 |
118.0 |
442.5 |
|
| DANHTO |
314.4 |
128.3 |
441.9 |
|
| TAHTO |
372.6 |
|
413.2 |
|
| RDX |
|
|
|
145.5 (145.6a) |
| HMX |
|
|
|
160.4 (160.4a) |
Table 4 The h50 values of HTO, TNHTO, ADNHTO, DANHTO, TAHTO, and TNT
| Compound |
h50 (cm) |
| Experimental values from ref. 41. Experimental values from ref. 36. |
| s-heptazine |
107 |
| HTO |
57 |
| TNHTO |
10 |
| ADNHTO |
30 |
| DANHTO |
88 |
| TAHTO |
161 |
| RDX |
30 (26a) |
| HMX |
26 (29a) |
| PA |
67 (64a) |
| TNT |
100 (98a) |
| LLM-105 |
110 (117b) |
Overall, symmetrically introducing six N-oxides into s-heptazine can obviously enhance its HOF, density, and explosive performance but will decrease the thermal stability and increase the sensitivity to some extent.
3.2 Structure–properties relationships
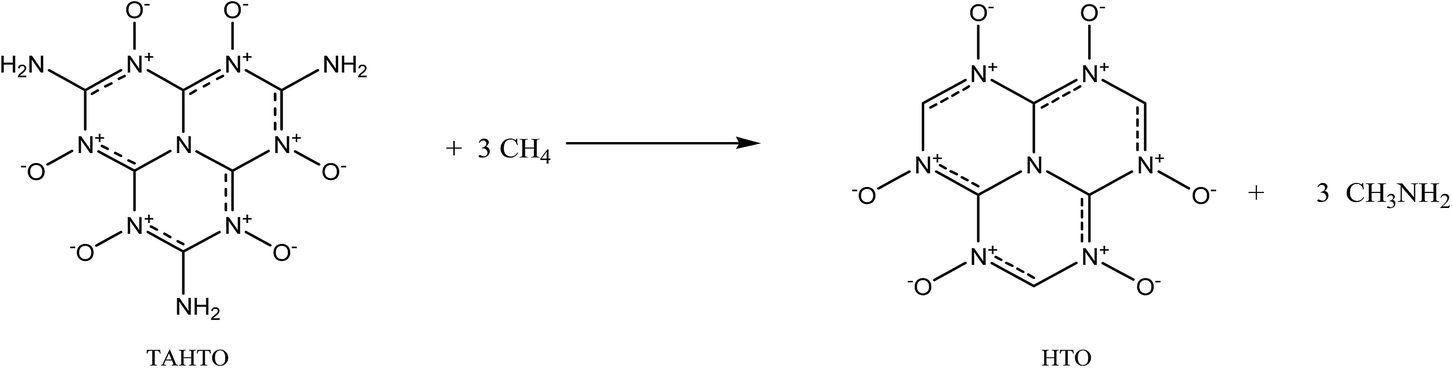
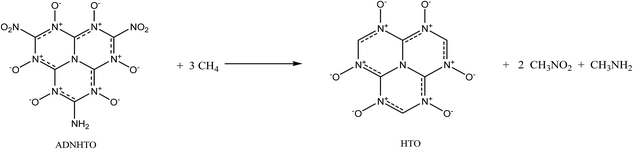
In this section, we turn to investigate the structure–property relationships for the HTO derivatives by replacing the three hydrogen atoms of HTO with the amino and/or nitro groups. First, a new compound trinitroheptazine-1,3,4,6,7,9-hexaoxides (TNHTO, Fig. 1) is obtained by substituting the three hydrogen atoms in HTO by nitro groups. The optimized structure of TNHTO is displayed in Fig. 2. It is found that all the atoms are coplanar approximately except for the six oxygen atoms in the three nitro groups, which are out of the network plane constructed by the rest atoms. This shows that the planarity of TNHTO is much worse than that of HTO and the conjugation effect in the former is weaker than that in the later. From Table 1, it is seen that the C–NO2 bond lengths in TNHTO are in line with the length of normal C–N single bond and are obviously longer than those of all the C–N bonds in the ring, indicating that the nitro groups hardly participate in the conjugation of the ring. Then, two other new compounds aminodinitroheptazine-1,3,4,6,7,9-hexaoxides (ADNHTO, Fig. 1) and diaminonitroheptazine-1,3,4,6,7,9-hexaoxides (DANHTO, Fig. 1) are designed by replacing the three hydrogen atoms in HTO by two nitro groups and one amino group and one nitro group and two amino groups, respectively. Their optimized structures in Fig. 2 indicate that the oxygen atoms in nitro groups are completely out of the approximate net plane, which is the same as that of TNHTO. It is seen in Table 1 that the bond lengths of the C–NO2 and C–NH2 bonds are between the normal C–N single bond length and CN double bond length but close to the C–N bond length, indicating that the nitro groups do not participate in the conjugate system, while the amino groups take part in the conjugation of the big fused ring well. The last compound triaminoheptazine-1,3,4,6,7,9-hexaoxides (TAHTO, Fig. 1) is gained by substituting the three hydrogen atoms in HTO with three amino groups. Its optimized structure in Fig. 2 displays that all the atoms in TAHTO are coplanar, indicating that it is a big conjugated planar molecule. Also, it is seen in Table 1 that the bond lengths of all the C–N bonds and N–O bonds are between the common C–N (N–O) single bond length and C–N (N–O) double bond length. In all, the introduction of nitro groups into HTO can damage the structure planarity and weaken the conjugation of the molecule; moreover, this is enhanced with the increment of the numbers of nitro groups, which may suggest that their thermal stability would decrease in the same sequence, while the substitution of the amino groups just plays the opposite role. In addition, all five designed compounds HTO, TNHTO, ADNHTO, DANHTO, and TAHTO are planar or approximate planar molecules, indicating that their properties can be suitably estimated by the Politzer's methods.27,31
Table 2 lists the calculated solid-phase HOFs, oxygen balance (OB), ρ, D, and P of HTO, TNHTO, ADNHTO, DANHTO, and TAHTO. Fig. 3 compares the effects of different substitution of nitro and amino groups on the energetic properties of the title compounds. All the substituted derivatives have lower HOFs but higher ρ, D, and P than HTO, indicating that the substitutions of nitro groups and amino groups decrease the HOF but enhance the ρ, D, and P of HTO. The HOF, OB, D, and P of the derivatives decrease in the sequence: TNHTO > ADNHTO > DANHTO > TAHTO, suggesting that increasing the numbers of nitro groups is an effective method to improve the detonation performance. Since introducing the amino groups into HTO is very helpful for generating extensive intramolecular and intermolecular hydrogen bonds that can increase the densities, the densities of DANHTO, TAHTO, and ADNHTO are close each other and are higher than that of TNHTO. In all, incorporating nitro groups or amino groups into the HTO system can obviously improve its detonation performance.
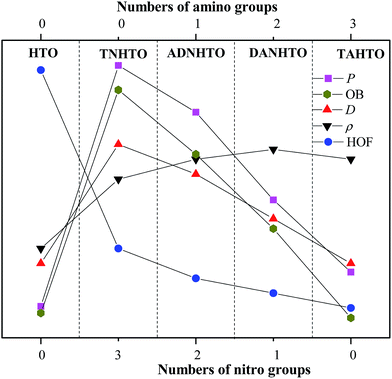 |
| | Fig. 3 A comparison of the effects of different substitution on the solid-phase HOFs, OB, ρ, D, and P of the title compounds. | |
Fig. 4 displays a comparison of the ρ, D, and P of HTO, TNHTO, ADNHTO, DANHTO, TAHTO, HMX, and two most powerful high explosives that have been synthesized until now 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20) and 1,2,3,4,5,6,7,8-octanitrocubane (ONC). All the five designed new explosives have higher ρ, D, and P than HMX. TNHTO, ADNHTO, DANHTO, and TAHTO have higher ρ than CL-20 and ONC. The D and P of HTO and TAHTO are slightly lower than those of CL-20 but are comparable with those of ONC. DANHTO, ADNHTO, and TNHTO have higher D and P than CL-20. Overall, all the designed explosives have outstanding detonation performance. However, for an ideal high explosive, both high energy and low sensitivity are required. Thus, further investigations should be done on studying their stability and sensitivity.
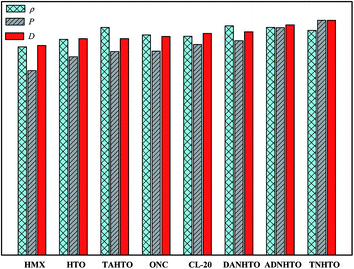 |
| | Fig. 4 A comparison of ρ, D, and P of HTO, TNHTO, ADNHTO, DANHTO, TAHTO, HMX, CL-20, and ONC. | |
The calculated BDE of the relatively weak bonds of HTO, TNHTO, ADNHTO, DANHTO, TAHTO, RDX, and HMX are listed in Table 3. Our calculated BDE values of RDX and HMX are very close to other calculated results. All the substituted derivatives have lower BDE values than HTO, indicating that the former have worse thermal stability than the later. The BDE values of the derivatives decrease in the sequence TAHTO > DANHTO > ADNHTO > TNHTO, suggesting that their thermal stability decrease in the same order. Therefore, increasing the numbers of amino groups can improve the thermal stability of the title compounds. This is in agreement with the results observed from the variation of the structure planarity with the numbers of amino groups or nitro groups. The BDE values of the C–NO2 bond are lower than those of other bonds for TNHTO, ADNHTO, and DANHTO, suggesting that the C–NO2 bond cleavage is an initial decomposition step. For TAHTO, the BDE of the (C–N)ring bond is the lowest among other bonds, indicating that the ring opening is an initial decomposition step of TAHTO. The lowest BDE values of TNHTO, ADNHTO, and DANHTO are lower than those of RDX and HMX, while for HTO and TAHTO, the situation is opposite, showing that the former three compounds have worse thermal stability, but the latter two one have better thermal stability compared with RDX and HMX. In addition, the lowest BDE values of TNHTO and ADNHTO are only around 110 kJ mol−1. Their low BDE values indicate that they are very sensitive and have poor thermal stability.
Table 4 lists the calculated h50 values of TNHTO, ADNHTO, DANHTO, TAHTO, RDX,41 HMX,41 PA,41 TNT,41 and LLM-105.36 The calculated h50 values of RDX, HMX, PA, TNT, and LLM-105 are very close to their experimental results, respectively. It is found that h50 increases decreases in the sequence of TNHTO, ADNHTO, DANHTO and TAHTO. This means that the sensitivity decreases with the same order. Thus, increasing the numbers of amino groups is helpful for reducing the sensitivity. Previous studies reported40,41 that the electrostatic potential (ESP) is related to the impact sensitivity of the energetic materials, and the stability can be expressed as a function of the imbalance between positive and negative regions. In the N–O systems, the regions of stronger positive potential are concentrated on the nitrogen atom and lead to the atypical imbalance which causes high impact sensitivity. It is seen in Fig. 5 that the positive potential around the nitrogen atoms of the N–O systems becomes less and less in the order of TNHTO, TNHTO, ADNHTO, HTO, DANHTO, TAHTO, indicating that their sensitivity reduces with the same sequence, which agrees with the results inferred form the changing trend of h50 values.
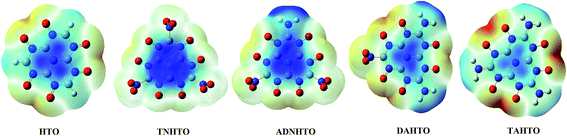 |
| | Fig. 5 The electrostatic potential [0.001 electron bohr−3 isosurface, color coding: from yellow (negative) to blue (positive)] of HTO, TNHTO, ADNHTO, DANHTO, and TAHTO. | |
Fig. 6 displays a comparison of h50 of HTO, TNHTO, ADNHTO, DANHTO, TAHTO, HMX, ONC, and one widely used insensitive explosive TNT. TNHTO has lower h50 than HMX, indicating that it is very sensitive, which may be caused by the bad structure planarity, weak conjugation, and the formation of no hydrogen-bonding. ADNHTO has comparable h50 with HMX, suggesting that it is as sensitive as HMX. The h50 of HTO and DANHTO are higher than that of ONC but lower than that of TNT, indicating that they are more insensitive than ONC but more sensitive than TNT. TAHTO has obvious higher h50 than TNT, indicating that it is a very insensitive explosive, which may be since it is a well planar molecule with a lot of hydrogen bonds generated by the N-oxides and amino groups and there are no obvious weak bonds in it like the N–NO2 and C–NO2 bonds. In all, based on the detonation performance and sensitivity, all the designed new molecules are more powerful and insensitive than HMX except for TNHTO. DANHTO has higher explosive performance than CL-20 and its sensitivity is close to TNT, while TAHTO has comparable detonation performance with ONC and lower sensitivity than TNT, showing that they are two outstanding high explosives with high energy and low sensitivity. Therefore, properly incorporating N-oxides, amino groups, and nitro groups into s-heptazine are helpful for improving its detonation performance. This strategy may be used to design and develop other new energetic materials.
 |
| | Fig. 6 A comparison of h50 of HTO, TNHTO, ADNHTO, DANHTO, TAHTO, HMX, ONC, and TNT. | |
Referring to the synthesis of a similar compound LLM-105, here we suppose the synthetic routes for the five designed energetic materials. HTO may be obtained by using appropriate oxidizers like peroxyformic acid to oxidize s-heptazine. For TAHTO, the first step is to synthesize trichloroheptazine, the next is convert it into triaminoheptazine, and the final is to oxidize triaminoheptazine to TAHTO step by step by employing suitable oxidizing agents. For the rest three compounds, starting from monochloroheptazine, dichloroheptazine, and trichloroheptazine, aminodinitroheptazine (ADNH), diaminonitroheptazine (DANH), and tridinitroheptazine (TNH) may be synthesized, respectively. Then, strong oxidants are used to oxidize ADNH, DANH, and TNH to obtain ADNHTO, DANHTO, and TNHTO, respectively. Since all of them have six N-oxides and some of them have amino and nitro groups, it is necessary to synthesize stronger oxidant agents and develop more efficient and suitable oxidation methods than existing ones. Due to the unique structures of the five designed compounds, further studies are needed.
3.3 Electronic structure
Table 5 lists the calculated HOMO–LUMO gap (ΔE), ionization potential (IP), and electron affinity (EA) of HTO, TNHTO, ADNHTO, DANHTO, and TAHTO. All the four substituted derivatives have higher ΔE than HTO, indicating that the electron transition from HOMO to LUMO in the former is more difficult than that in the later. Among the five designed compounds, TNHTO and TAHTO have the highest IP and EA, respectively, suggesting that it is most difficult to create a hole and accept an electron in TNHTO and TAHTO, respectively. Their ΔE values enhance with the increment of the numbers of amino groups, indicating that incorporating the amino groups into the systems makes the electron transitions more difficult. However, the case for IP and EA is just the opposite, suggesting that introducing the amino groups into the molecules makes the loss of electron difficult but does the gain of electron easy.
Table 5 Calculated HOMO–LUMO gap (ΔE, eV), ionization potential (IP, eV), and electron affinity (EA, eV) of HTO, TNHTO, ADNHTO, DANHTO, and TAHTO
| Compound |
ΔE |
IP |
EA |
| HTO |
1.55 |
7.34 |
3.00 |
| TNHTO |
1.63 |
8.11 |
4.08 |
| ADNHTO |
1.66 |
7.55 |
3.76 |
| DANHTO |
1.89 |
7.05 |
2.98 |
| TAHTO |
1.93 |
6.41 |
2.07 |
3.4 Spectral properties
Fig. 7 depicts the calculated IR spectrums of HTO, TNHTO, ADNHTO, DANHTO, and TAHTO. Obviously, the five molecules have similar IR spectra. The strongest peaks at around 1600 cm−1 correspond to the NO asymmetric stretch of nitro groups and the N–O bonds linked to ring. The strong peaks at around 1200 cm−1 are associated with the C–N stretch and the NO symmetric stretch motion of nitro groups and the N–O bonds linked to the ring. The weak peaks at over 3000 cm−1 correspond to the C–H or N–H stretch modes. Fig. 8 displays the calculated UV-VIS spectrums of HTO, TNHTO, ADNHTO, DANHTO, and TAHTO. All of them have strong absorption in the region of visible light and only TAHTO can absorb ultraviolet light. In the VIS-light region, the strongest peaks at around 427 nm (TAHTO), 545 nm (DANHTO), 592 nm (HTO), 596 nm (ADNHTO), and 664 nm (TNHTO) are located in the region of purple light, green light, yellow light, yellow light, and red light, respectively, indicating that they probably are yellow-green, red-purple, blue, blue and blue-green compounds, respectively.
 |
| | Fig. 7 The calculated IR spectrums of HTO, TNHTO, ADNHTO, DANHTO, and TAHTO. | |
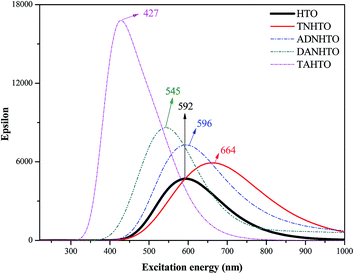 |
| | Fig. 8 The calculated UV-VIS (in dimethylsulfoxide solution) spectrums of HTO, TNHTO, ADNHTO, DANHTO, and TAHTO. | |
4 Conclusions
In this work, we present a strategy to design five new high explosives HTO, TNHTO, ADNHTO, DANHTO, and TAHTO by introducing N-oxides, nitro and amino groups into s-heptazine. Their energetic properties and sensitivity are estimated by using DFT and compared with some famous explosives like CL-20, ONC, and TNT. It is found that all the five explosives have much higher densities, HOFs, and detonation performance than s-heptazine. Increasing the numbers of nitro groups can obviously improve the HOF, OB, D, and P, while enhancing the numbers of amino groups are very helpful for improving the structure planarity, conjugation effect, and thermal stability and reducing the sensitivity. All the designed new compounds are more powerful and insensitive than HMX except for TNHTO, which has higher energy than CL-20 but is more sensitive than HMX. DANHTO has better explosive performance than CL-20 and its sensitivity is close to TNT, while TAHTO has comparable detonation performance with ONC and lower sensitivity than TNT, indicating that they are outstanding high explosives with high energy and low sensitivity. This indicates that properly incorporating N-oxides, amino groups and nitro groups into s-heptazine can generate new explosives with excellent performance. This strategy may be used to design and develop other new energetic materials.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 21273115) and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions. Q. Wu would like to thank the Innovation Project for Postgraduates in Universities of Jiangsu Province (Grant no. CXZZ13_0199) for partial financial support.
References
- R. S. Hosmane, M. A. Rossman and N. J. Leonard, J. Am. Chem. Soc., 1982, 104, 5497–5499 CrossRef CAS
 .
. - W. X. Zhang, N. B. Wong, G. Zhou, X. Q. Liang, J. S. Li and A. M. Tian, New J. Chem., 2004, 28, 275–283 RSC .
- W. X. Zhang, N. B. Wong, W. K. Li and A. M. Tian, J. Phys. Chem. A, 2004, 108, 11721–11727 CrossRef .
- E. Kroke, M. Schwartz, E. Horath-Bordon, P. Kroll, B. Noll and A. D. Norman, New J. Chem., 2002, 26, 508–512 RSC .
- B. Jürgens, E. Irran, J. Senker, P. Kroll, H. Müller and W. Schnick, J. Am. Chem. Soc., 2003, 125, 10288–10300 CrossRef PubMed .
- D. R. Miller, D. C. Swenson and E. G. Gillan, J. Am. Chem. Soc., 2004, 126, 5372–5373 CrossRef CAS PubMed .
- R. Meyer, J. Köhler and A. Homburg, Explosives, Wiley-VCH, Verlag GmbH, Weinheim, 2007 Search PubMed .
- T. D. Tran, P. F. Pagoria, D. M. Hoffman, J. L. Cutting, R. S. Lee and R. L. Simpson, the 33rd International Annual Conference of ICT, Karlsruhe, Germany, 2002 Search PubMed .
- R. A. Hollins, L. H. Merwin, R. A. Nissan, W. S. Wilson and R. Gilardi, J. Heterocycl. Chem., 1996, 33, 895–904 CrossRef CAS .
- Z. P. Demko and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2110–2113 CrossRef CAS .
- H. M. Xiao and Z. X. Chen, The Modern Theory for Tetrazole Chemistry, Science Press, Beijing, 1st edn, 2000 Search PubMed .
- X. H. Ju, Y. M. Li and H. M. Xiao, J. Phys. Chem. A, 2005, 109, 934–938 CrossRef CAS PubMed .
- W. H. Zhu, C. C. Zhang, T. Wei and H. M. Xiao, J. Comput. Chem., 2011, 32, 2298–2312 CrossRef CAS PubMed .
- J. J. Zhang, H. C. Du, F. Wang, X. D. Gong and Y. S. Huang, J. Phys. Chem. A, 2011, 115, 6617–6621 CrossRef CAS PubMed .
- F. Wang, H. C. Du, J. H. Zhang and X. D. Gong, J. Phys. Chem. A, 2011, 115, 11788–11795 CrossRef CAS PubMed .
- Q. Wu, Y. Pan, X. L. Xia, Y. L. Shao, W. H. Zhua and H. M. Xiao, Struct. Chem., 2013, 24, 1579–1590 CrossRef CAS PubMed .
- Q. Wu, W. H. Zhu and H. M. Xiao, J. Chem. Eng. Data, 2013, 58, 2748–2762 CrossRef CAS .
- Q. Wu, W. H. Zhu and H. M. Xiao, RSC Adv., 2014, 4, 3789–3797 RSC .
- Q. Wu, W. H. Zhu and H. M. Xiao, J. Mater. Chem. A, 2014, 2, 13006–13015 CAS .
- P. W. Atkins, Physical chemistry, Oxford University Press, Oxford, 1982 Search PubMed .
- P. Politzer, J. S. Murray, M. E. Grice, M. DeSalvo and E. Miller, Mol. Phys., 1997, 91, 923–928 CrossRef CAS .
- P. Politzer and J. S. Murray, Cent. Eur. J. Energ. Mater., 2011, 8, 209–220 CAS .
- E. F. C. Byrd and B. M. Rice, J. Phys. Chem. A, 2006, 110, 1005–1013 CrossRef CAS PubMed .
- F. A. Bulat, A. Toro-Labbe, T. Brinck, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 1679–1691 CrossRef CAS PubMed .
- M. Jaidann, S. Roy, H. Abou-Rachid and L. S. Lussier, J. Hazard. Mater., 2010, 176, 165–173 CrossRef CAS PubMed .
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS PubMed .
- P. Politzer, J. Martinez, J. S. Murray, M. C. Concha and A. Toro-Labbé, Mol. Phys., 2009, 107, 2095–2101 CrossRef CAS .
- S. W. Benson, Thermochemical kinetic, Wiley-Interscience, New York, 2nd edn, 1976 Search PubMed .
- I. Mills, T. Cvitas, K. Homann, N. Kallay and K. Kuchitsu, Quantities, Units, and Symbols in Physical Chemistry, Blackwell Scientific Publications, Oxford, 1988 Search PubMed .
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed .
- M. Pospíšil, P. Vávra, M. C. Concha, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 895–901 CrossRef PubMed .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. AlLaham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Pittsburgh, PA, 2003 Search PubMed .
- A. F. Wells, Structural Inorganic Chemistry, Oxford University Press, Oxford, 3rd edn, 1962 Search PubMed .
- V. I. Pepekin, Y. N. Matyushin and Y. A. Lebedev, Russ. Chem. Bull., 1974, 23, 1707–1710 CrossRef .
- H. S. Dong and F. F. Zhou, High Energy Detonators and Correlative Performances, Science Press, Beijing, 1989 Search PubMed .
- R. D. Gilardi and R. J. Butcher, Acta Crystallogr., 2001, 57, 657–658 Search PubMed .
- H. X. Ma, J.
R. Song, F. Q. Zhao, H. X. Gao and R. Z. Hu, Chin. J. Chem., 2008, 26, 1998–2002 Search PubMed .
- T. D. Tran, P. F. Pagoria, D. M. Hoffman, J. L. Cutting, R. S. Lee and R. S. Simpson, in 12th Detonation Symposium, San Diego, 2002 Search PubMed .
- T. Wei, W. H. Zhu, X. W. Zhang, Y. F. Li and H. M. Xiao, J. Phys. Chem. A, 2009, 113, 9404–9412 CrossRef CAS PubMed .
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS .
- B. M. Rice and J. J. Hare, J. Phys. Chem. A, 2002, 106, 1770–1783 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.