
Bonding in exohedral metal–fullerene cationic complexes
Maitreyi Robledoa,
Néstor F. Aguirrea,
Sergio Díaz-Tendero*a,
Fernando Martínabc and
Manuel Alcamíab
aDepartamento de Química, Módulo 13, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: sergio.diaztendero@uam.es
bInstituto Madrileño de Estudios Avanzados en Nanociencias (IMDEA-Nanociencia), Cantoblanco, 28049 Madrid, Spain
cCondensed Matter Physics Center (IFIMAC), Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 3rd October 2014
Abstract
We present a systematic theoretical study of the exohedral interaction between singly positively charged metal (M) atoms and the C60 fullerene in [M–C60]+ complexes. Calculations have been carried out by means of the density functional theory. We have considered alkali (Li, Na and K), alkaline-earth (Be, Mg and Ca) and first period transition metals (Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu and Zn) interacting with C60 in different positions: on top of a ring (pentagon or hexagon), a bond (hexagon–hexagon or hexagon–pentagon) and a C atom. A detailed topological analysis of the electronic density reveals metal–C60 exohedral interaction of the ion-induced dipole type, with the positive charge localized on the metal atom and with increasing covalent character for the heavier transition metals. An energy decomposition analysis allows us to quantify the different contributions to the bonding in each complex, being dominant the polarization one. A simple ion-induced dipole model explains the main features of the interaction.
1 Introduction
The discovery of the C60 fullerene in 1985 (ref. 1) gave birth to a new era of carbon chemistry and, in particular, to the possibility to synthesize carbon compound derivatives with unusual physical and chemical properties relevant for industrial applications and nanotechnology. In this context, only a few years later, it was demonstrated the ability of C60 to form molecular solids in a close-packing structure.2 In these solids, the C60 molecules are not completely stuck; therefore, there is still space in between the cages that can be filled with other atoms, for instance metal atoms. This task was accomplished with the synthesis of the first alkali-doped fullerene solids:3,4 the so called fullerides [MxC60; M = Li, Na, K, Cs, Rb; x = 1–6]. From these pioneering studies, an increasing activity on metal doped C60 solids gave rise to several works in which fulleride properties were exposed, including, e.g., superconductivity in RbxC60 species,5 elucidation of structural properties in NaxCsyC60 superconductors,6 phase diagrams of alkali-metal fullerides with Rb and Cs,7 C60 thin film crystal growth on layered materials8 or superconductivity in mixed sodium and mixed lithium alkali compounds [Na2MC60 (M = K, Rb or Cs) and Li2CsC60].9 The stability and superconductivity properties of these compounds mainly rely on the charge transfer between the metal atoms and the fullerenes.Doping of single C60 molecules was first achieved by Martin et al.10 using alkali metals. In this work, it was observed a metal clustering process over the fullerene surface yielding a mixture of carbon–metal complexes: (C60)nMx (n = 1, 2, 3; 1 ≤ x < 150; M = Li, Na, and K). Theoretical studies have evaluated the structural and electronic properties of NanC60 and LinC60 (n ≤ 12) clusters within density functional theory11 and ab initio molecular dynamics simulations have shown the possibility that Li12C60 might exist as a stable cluster.12 Other theoretical studies have employed analytical model potentials based on polarization energies to understand the endohedral and exohedral adsorption of alkali ions in C60.13,14
Though the first choice in doping single C60 molecules (and solids) was using alkali metals, alkaline-earth metals have also been employed. In the mass spectra experiments of Zimmermann et al.,15 multilayer metal coverage of the fullerene molecules was proposed in C60 and C70 coating with alkaline-earth metals; they observed magic number species such as C60M32 (M = Ca, Ba and Sr).
Transition metals have been usually placed inside the fullerene cage, leading to the so-called endofullerenes (see e.g. ref. 16). These metals have been much less used to coat the fullerene surface. Mass spectrometric studies17 have shown that transition metals show a distinct preference to form stable droplet structures on the surface of the carbon cage instead of coating the whole fullerene outer shell. In addition, theoretical simulations on C60Ti62 (ref. 18) showed that the structure is stabilized by mixed ionic and covalent bonding between the 12 Ti atoms situated above the C pentagons and the fullerene, and the presence of a covalently bonded network of 50 Ti atoms around. In general, transition metals prefer to form clusters on the fullerene surface because the binding energy between the metal atoms and the carbon cage is lower than in the metal bulk.19 Previous works also focussed on metal-buckybowls binding properties.20–22
From a technological point of view, it has been found that metal doped carbon structures can lead to interesting applications for hydrogen storage devices.23–29 Some of the studied systems in this field are Li12C60, which is able to bind up to 60 hydrogen molecules30 or Ca32C60,31 which presents a stronger H2 adsorption energy than the former, and thus hydrogen desorption takes place at higher temperatures. Decorating carbon structures with transition metals has also been proposed as an alternative for efficient hydrogen storage.32 Although the clustering of these metals on the fullerene surface, minimizes the ability of the carbon cage to accumulate hydrogen in high quantities, it has been shown that metallofullerenes as C60Sc12 are able to bind up to 5 H2 molecules per Sc atom,33 and boron substituted fullerenes as C48B12 can further increase this H2 storage capacity.33
Increasing the total charge of the cluster can be used as a tool to study these complexes, for instances the study of charged fullerenes doped with atoms in He droplets and using high-resolution mass spectrometry allow the identification of specific adsorption sites of single atoms or molecules in charged C60 and C70 fullerenes.34,35 Similar techniques could be also employed to study the charged metal complexes considered in the present work. Also important, the increase of the charge of the system enhances some important properties. For instance, in the case of a neutral Li atom is able to bind just one H atom while a Li+ ion can bind up to 12 H atoms,36 which points out the importance of considering charged metal doped fullerenes in this field. As in the case of Li,36 C60 complexes with charged transition metals could also enhance the number of H2 molecules that can bind to the metal. To the best of our knowledge this possibility has not been investigated. This is the objective of the present work: to study in a systematic way how the properties of metal–fullerene bond vary with the nature of the metal. Understanding the nature of the bonding in metal–fullerene charged complexes is relevant for future applications based in these complexes.
In this work we present a systematic theoretical study on the exohedral interaction between a single alkali, alkaline-earth or transition metal cation and the C60 fullerene. The nature of the chemical bond is characterized from the analysis of the electronic density and localized charge distribution, in combination with an energy decomposition analysis.
If we exclude the case of beryllium cation, which has an anomalous behaviour previously observed,37–39 the metal–fullerene interaction is dominantly ionic in the case of alkali and alkali-earth metals and it has an increasing covalent character as one moves along the first period of transition metals. This suggests that, in addition to alkalis and alkali-earths (excluding Be), the lighter elements in the first transition period could also be used to build [MnC60]q+ complexes with a substantial capacity to store hydrogen.
2 Computational details
We have performed self-consistent field calculations in the framework of the Density Functional Theory (DFT). In particular, our calculations were carried out with the hybrid B3LYP functional, which combines the Becke's three parameter non-local hybrid exchange potential40 with the non-local gradient corrected correlation functional of Lee, Yang and Parr.41 This functional has been used in combination with the Pople 6-31G(d) basis set for the geometry optimization in a first step towards finding the most stable isomers: for each [C60M]+ complex studied different geometries and spin multiplicity were considered. We have performed the geometry optimizations without symmetry restrictions. Harmonic vibrational frequencies have been also evaluated at the same B3LYP/6-31G(d) level to confirm that the calculated geometry effectively corresponds to a minimum or a saddle point (first order transition state – TS) in the potential energy surface (PES) and to compute the Zero Point Energy (ZPE) correction. Once we have located the most stable isomer for each complex Single Point Energy (SPE) calculations at the B3LYP/6-311+G(d) level of theory were carried out in order to obtain more accurate values of the metal–fullerene interaction energy. The Basis Set Superposition Error (BSSE) has been also calculated at this level of theory. All these calculations were carried out with the Gaussian 09 program package.42In most of the systems under study we are dealing with electronic open shells and we are thus performing unrestricted DFT calculations (UDFT). The UDFT wavefunction is no longer an eigenfunction of the total spin, S2 and spurious results due to the error known as spin contamination can be obtained. We have checked the mean value of 〈S2〉 for all the [C60M]+ complexes studied to verify the existence of this error. In the worst cases ([C60Ni]+ and [C60Fe]+) the obtained value is only 2% higher than the expected one. Thus, we can conclude that the computed wave functions are not spin contaminated.
We have analyzed the electronic density properties by means of the Quantum Theory of Atoms In Molecules (QTAIM) by R. Bader.43,44 The corresponding calculations have been carried out using the AIMALL (version 11.12.19) program package.45
We have also studied the type of interaction in the framework of the Natural Bond Orbital (NBO) approach46–48 as implemented in the Gaussian package.49 An atomic charge analysis has been analyzed in the framework of the Mulliken population analysis.50 It is well known that the inconsistencies obtained in the Mulliken results are a consequence of its basis dependence; hence, in order to avoid possible spurious results we have also studied the atomic charges employing the NBO method (Natural Population Analysis – NPA), which has already given good results for similar species with silicon–metal atoms interactions.51,52
The optimization of the fullerene cage leads to a length of 1.39 Å for the bond between two hexagons (hh bond) and 1.45 Å for the one between a pentagon and a hexagon (hp bond). These results are in good agreement with previous theoretical works, e.g. Rhh = 1.38 Å, Rhp = 1.43 Å,53 Rhh = 1.40 Å, Rhp = 1.46 Å,11 and with electron diffraction measurements Rhh = 1.40 Å, Rhp = 1.45 Å.54
Energy decomposition analysis within the localized molecular orbital method (LMO-EDA)55 was carried out with the GAMESS package.56,57 For the complexes here considered, this method is computationally very expensive on all-electron basis set calculations within the 6-311+G(d) basis, in particular in the case of open shell systems. We have thus used Effective Core Pseudo-potentials (ECP) to overcome the problem. Additionally, in order to keep invariant the basis set we removed the first contracted shells in a consistent way, including the number of electrons associated with the ECP. Reoptimization of the basis sets for being used with the new ECP was not necessary: we checked for Cu and Li complexes than the interaction energies computed with an all electron calculations and with the ECP one differ in less than 8%. On the other hand, the LMO-EDA components for these two systems, are well reproduced using the reduced basis sets. In all cases the interaction energy was computing considering two monomers: the neutral C60 with singlet spin multiplicity and the singly charged metal atom with the corresponding multiplicity. The super-molecule is also singly charged and shows the same spin multiplicity of the metal cation. The notation ECP/basis represents the pair ECP combined with the basis set used for each atom. For example, ECP10MWB/6-311+G(d) means that the pseudo-potential ECP10MDF and the basis set 6-311+G(d) were used to represent the atom. If the ECP was taken from the web page of Stuttgart/Cologne group,58 the number of electrons is included into its label (in this example the number of core electrons is 10). The basis sets were taken from the Gaussian software package.42 For example in Co the 6-311+G(d) basis have the following contraction scheme (15s,11p,6d,1f) → [10s,7p,4d,1f]. So, according with the number of core electrons associated with the ECP and the core electronic configuration 1s22s22p6, the two first “s” and one “p” contractions were removed from the basis set, i.e. (8s,6p,6d,1f) → [8s,6p,4d,1f]. This strategy was followed for the all the atoms considered in this work (the ECPs and basis sets employed are listed in Table 1). The counterpoise (CP) method59 was used to estimate the basis set superposition error (BSSE) for the total interaction energies and the corresponding components. The result including the BSSE corrections is shown as error bars in the figures. In all cases, the BSSE is lower than 4 kcal mol−1, except for the repulsion component in Ti complex (∼8 kcal mol−1). This shows that the level of theory here employed for the LMO-EDA calculations (either with or without the CP correction) is accurate enough to investigate the intermolecular interactions of this kind of complexes.
| Atom | ECP/basis set | Core | |
|---|---|---|---|
| Electrons | Config. | ||
| C | ECP2MWB/6-31G(d) | 2 | 1s2 |
| Li | 6-311+G(d) | 0 | |
| Na | CRENBL/6-311+G(d) | 2 | 1s2 |
| K | ECP10MWB/6-311+G(d) | 2 | 1s2 |
| Be | 6-311+G(d) | 0 | |
| Mg | CRENBL/6-311+G(d) | 2 | 1s2 |
| Ca | ECP10MWB/6-311+G(d) | 10 | 1s22s22p6 |
| [Sc–Zn] | ECP10MWB/6-311+G(d) | 10 | 1s22s22p6 |
We carried out calculations of the polarizability for each metal cation at the CCSD(T) level (coupled cluster method including all single and double excitations and a quasi-perturbative treatment of connected triple excitations) with the MOLPRO (2010.1 version) package,60 by using the polarization consistent basis sets developed by Jensen,61–63 specifically that known as aug-pc-4. The polarizabilities have been obtained as energy derivatives by the finite difference approximation. We kept invariant the same electronic spin configuration that the obtained in the metal–C60 complexes.
3 Results and discussion
Fig. 1a shows the five types of structures, with the metal atom interacting with the fullerene external surface, considered in this work: the hexagonal (h) and pentagonal (p) rings, the hh and hp bonds and on top of a carbon atom (C). Furthermore, several spin multiplicities have been also included in our calculations. The relative energies between the isomers obtained after geometry optimization, at the B3LYP/6-31G(d) level, are shown in Table 2.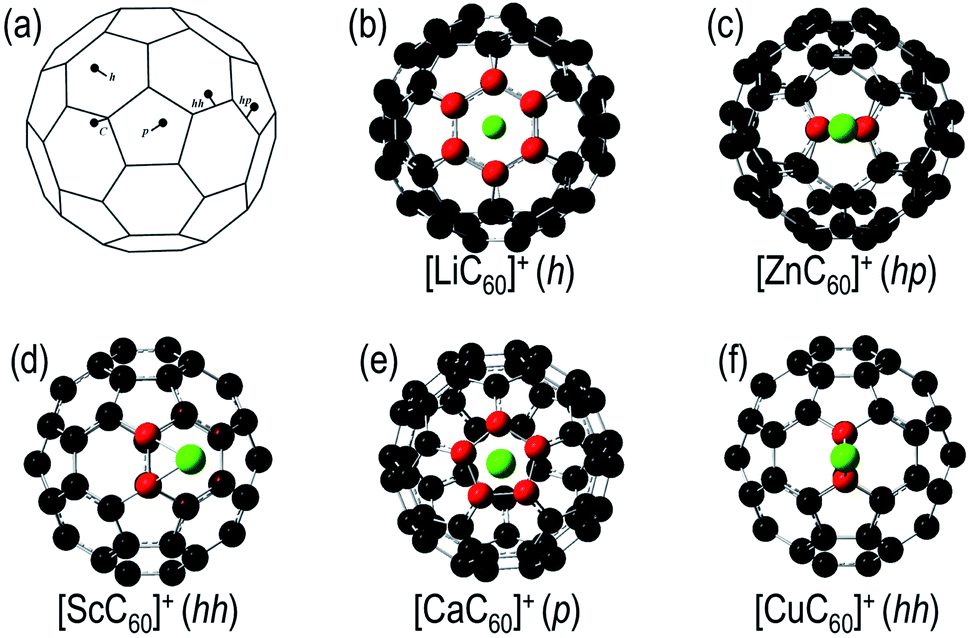 | ||
| Fig. 1 (a) The five types of initial geometries for [MC60]+, and the NPA charge distribution for (b) [LiC60]+, (c) [ZnC60]+, (d) [ScC60]+, (e) [CaC60]+, (f) [CuC60]+. Red atoms denote negative charge, green atoms positive charge and black atoms neutral (NPA performed within the NBO analysis). | ||
| Multiplicity | Isomer | ΔE | |
|---|---|---|---|
| a Li+ values taken from ref. 65. | |||
| [LiC60]+a | Singlet | h | 0.00 |
| Singlet | hh* | 4.17 | |
| Singlet | p | 0.01 | |
| Singlet | hp* | 3.18 | |
| [NaC60]+ | Singlet | h | 0.00 |
| Singlet | hh* | 2.85 | |
| Singlet | p | 0.54 | |
| [KC60]+ | Singlet | h | 0.00 |
| Singlet | hh* | 1.99 | |
| Singlet | p | 1.25 | |
| [BeC60]+ | Doublet | p | 2.59 |
| Doublet | hp | 0.00 | |
| Doublet | C | 0.25 | |
| [MgC60]+ | Doublet | p | 0.18 |
| Doublet | hp | 0.00 | |
| Doublet | C | 0.64 | |
| [CaC60]+ | Doublet | h | 0.60 |
| Doublet | p | 0.00 | |
| [ScC60]+ | Singlet | hh | 0.00 |
| Singlet | hp | 11.12 | |
| [TiC60]+ | Doublet | hh | 11.87 |
| Quadruplet | h | 0.00 | |
| [VC60]+ | Triplet | hh | 11.24 |
| Quintuplet | h | 0.00 | |
| [CrC60]+ | Sextuplet | h | 1.00 |
| Sextuplet | hh | 0.00 | |
| Sextuplet | p | 0.65 | |
| Sextuplet | hp | 1.14 | |
| [MnC60]+ | Quintuplet | hh | 2.28 |
| Septuplet | h | 0.00 | |
| [FeC60]+ | Quadruplet | hh | 0.00 |
| [CoC60]+ | Triplet | h | 3.45 |
| Triplet | hh | 0.00 | |
| [NiC60]+ | Doublet | hh | 0.00 |
| Doublet | p | 20.71 | |
| Doublet | hp | 3.11 | |
| [CuC60]+ | Singlet | h | 0.42 |
| Singlet | hh | 0.00 | |
| Singlet | p | 0.16 | |
| Singlet | hp | 2.10 | |
| [ZnC60]+ | Doublet | h | 2.93 |
| Doublet | p | 0.87 | |
| Doublet | hp | 0.00 | |
| Doublet | C | 0.01 | |
In the alkali metal complexes, the most stable geometry corresponds to the h isomer with singlet spin multiplicity in all cases. This result is consistent with previous works on neutral11,64 and singly charged alkali cation species.53,65 The relative energy between the h and p isomers increases in the sequence [LiC60]+, [NaC60]+, [KC60]+, being both isomers degenerated in the Li case and with a difference of only 1.25 kcal mol−1 in the K one. As described in previous studies for Li+,65 the energy barriers connecting different minima are very low. For instance the transition state (TS) that connects two equivalent h isomers (structure hh) has a relative energy of 4.17 kcal mol−1 with respect to the most stable isomer (h). We have evaluated the same TS(hh) for [NaC60]+ and [KC60]+, and the energy decreases to 2.85 and 1.99 kcal mol−1 respectively (see Table 2). Thus, these complexes present a soft potential energy surface (PES), with less pronounced energy barriers for the larger alkali metals. The most stable isomer in singly charged alkaline-earth complexes has doublet spin multiplicity and involves a pentagonal ring: hp for [BeC60]+ and [MgC60]+, and p for [CaC60]+, nearly degenerated with the C, p and h structures respectively.
We have also studied the ten elements belonging to the first row of transition metals. Transition metal cations are characterized by an increasing occupancy of their d orbitals in the valence electronic shell as we move forward through the periodic table row (from Sc+ 4s23d0 to Zn+ 4s13d10). Our present calculations show that the lowest energy structure involves a hexagonal ring: hh isomer for [ScC60]+, [CrC60]+, [FeC60]+, [CoC60]+, [NiC60]+ and [CuC60]+; h isomer for [TiC60]+, [VC60]+ and [MnC60]+; and hp isomer for [ZnC60]+. We observe two trends for the first row of transition metals for the spin multiplicities: (i) in the first half of the row the spin multiplicity increases from Sc (singlet) to Mn (septuplet). The preference for the largest spin multiplicity is easily explained by Hund's rule: the most stable atomic state is the one showing the largest spin multiplicity in an open subshell, in this case the five degenerated 3d atomic orbitals of the metal. (ii) For the second half of the row, a clear preference for low spin multiplicities is observed, due to the fact that these metals have almost filled the 3d valence shell. The spin multiplicities obtained correspond to the most stable electronic state for the isolated metal cation,66 pointing out that the electronic structure of the metal is not much distorted when forming the complex. The only two exceptions are Sc and Fe. In this latter case Fe+ (4F) is nearly degenerate with the ground state (6F).
The optimized geometries of the studied species show that the larger the radius of the metal cation, the larger the distance between the metal and the fullerene. Furthermore, the geometry of the carbon cage is almost not distorted after metal attachment in a ring (h or p). However, when the metal atom is attached to a bond (hh or hp), the C–C distance is enlarged by ∼0.03–0.17 Å. The largest distortion is observed in the Sc case. In the most stable isomer of the [ScC60]+ complex, the metal atom is attached to the hh C–C bond, softly leaning to one of the h rings, and abruptly opening the hh bond from 1.39 Å in the bare C60 up to 1.56 Å in the complex (see Fig. 1d). Sc is one of the elements, together with other rare earth metals as lanthanum, that forms stable endofullerenes.16 As an example, in a previous theoretical study on Sc@C60,67 the most stable isomer shows the metal atom placed 1.17 Å away from the center of the carbon cage, trying to reach the carbon shell where strong bonds with the C atoms can be formed.
The metal fullerene adsorption energy, Eads, is given by:
| Eads = (EC60 + EM+) − E[MC60]+ | (1) |
 | ||
| Fig. 2 Adsorption energy (in kcal mol−1) for the [MC60]+ complexes computed with DFT (B3LYP): all electron calculations with the 6-311+G(d) basis in filled symbols with full line, and LMO-EDA(B3LYP) using effective core pseudo-potentials and mixed basis sets in open symbols dotted line (see Computational details). Error bars represent the effect of BSSE, upper and lower limits represent the adsorption energy with and without BSSE correction respectively. | ||
In order to get a comprehensive view of the metal–fullerene interaction in the [MC60]+ exohedral complexes, we have analyzed several aspects considering the most stable isomer for each metal. We have first studied the atomic charge localized on the metal atom within the Mulliken Population Analysis, qMulliken, and the Natural Bond Orbitals (NBO) approach (Natural Population Analysis – NPA), qNBO (see Fig. 3). In the alkali and alkaline earth complexes the charge localized on the metal atom increases in the series Li < Na < K and Be < Mg < Ca, being in all cases close to q = +1 a.u. (when the NPA charges are considered), i.e. the whole positive charge of the complex is mainly localized on the metal atom. We also appreciate partial negative charge distribution localized on the carbon atoms of the hexagonal ring of the fullerene involved in the interaction with the alkali atom (see [LiC60]+ as an example in Fig. 1b). For the alkaline-earth metals a localized negative charge is also observed in the carbon atoms of the hp bond ([BeC60]+ and [MgC60]+) or p ring ([CaC60]+, see Fig. 1e). The π electronic cloud is polarized towards the cation leading to a partial negative charge on the atoms taking part in the interaction. This feature is the clear indication of a very localized interaction between the metal cation and the fullerene surface, since other carbon atoms surrounding the binding site do not show negative charge and, therefore, do not take place directly in the interaction.
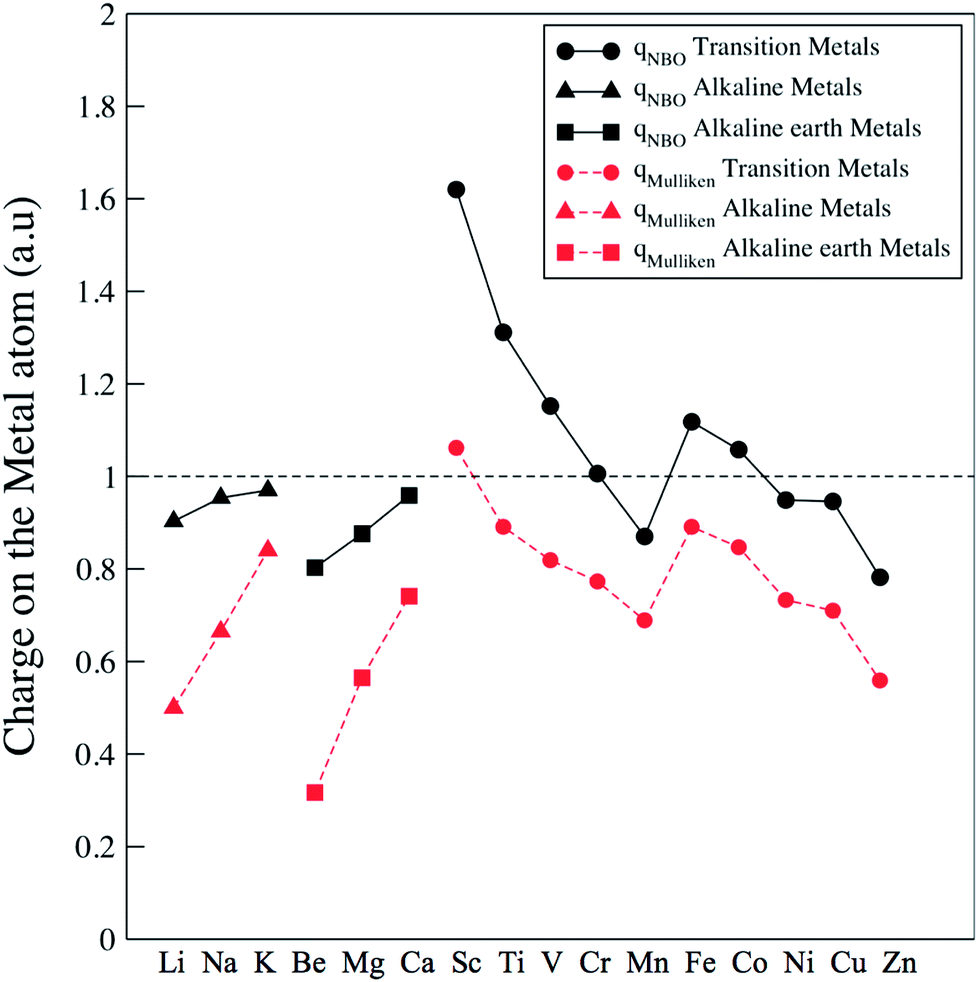 | ||
| Fig. 3 Charge localized on the metal cation of the [MC60]+ complexes computed with the Mulliken approach, qMulliken (dashed lines), and with the Natural Bonding Orbital theory qNBO (full lines), (circles transition, triangles-up alkali and squares alkaline-earth metals). | ||
The charge localized on the metal atom for the transition metal complexes shows a different behavior (see Fig. 3). Indeed in several cases the charge localized on the metal atom is larger than one; i.e. an excess of electronic density of the metal atoms is transferred to the carbon cage thus leading to a negatively-charged C60 cage. Also the trends observed for these metals can be clearly differentiated in two subgroups: from Sc to Mn and from Fe to Zn. In both subgroups the charge decreases in the corresponding sequence. Very localized interaction is also shown by the charge analysis of the carbon atoms (examples for Zn, Sc and Cu complexes are shown in Fig. 1). In the energy decomposition analysis (see below) we considered that the whole charge (+1) is on the metal cation; the adsorption energies obtained with this method follow the same trend as in all-electron DFT(B3LYP) calculations, which confirms indeed that the charge can be considered as located in the metal atom.
Further understanding of this behavior has been carried out on the basis of an exhaustive analysis of electron density properties. These results are summarized in Fig. 4 and 5 and in Table 3. Fig. 4 and 5 represent the so called molecular graphs of the most stable isomer for each [MC60]+ complex studied. They show the bond paths and the bond critical points (BCPs) in the fullerene–metal interaction region. These graphs include the contour map (2D plot) of the Laplacian of the electronic density, ∇2(ρ), and the contour map of the total energy density, H(ρ), as proposed by Cremer and Kraka.68 The analysis of both magnitudes provides complementary information.
 | ||
| Fig. 4 Molecular graphs of the most stable isomer of the [MC60]+ alkaline and alkaline earth metal complexes studied. Contour maps of the Laplacian of the electron density ∇2(ρ) and total electronic energy density H(ρ) are also shown. Positive values of H are denoted by solid black contours and negative values by dashed red contours. | ||
 | ||
| Fig. 5 Molecular graphs of the most stable isomer of the [MC60]+ transition metal complexes studied. Contour maps of the Laplacian of the electron density ∇2(ρ) and total electronic energy density H(ρ) are also shown. Positive values of H are denoted by solid black contours and negative values by dashed red contours. | ||
| ρ | ∇2ρ | G(ρ) | V(ρ) | H(ρ) | RC–M | Eads | |
|---|---|---|---|---|---|---|---|
| [LiC60]+ (h) | 0.13 | 0.07 | 0.0139 | −0.0110 | 0.0029 | 2.38 | 39.3 |
| [NaC60]+ (h) | 0.10 | 0.05 | 0.0097 | −0.0079 | 0.0018 | 2.74 | 22.8 |
| [KC60]+ (h) | 0.07 | 0.03 | 0.0062 | −0.0047 | 0.0014 | 3.19 | 15.3 |
| [BeC60]+ (hp) | 0.05 | 0.18 | 0.0605 | −0.0749 | 0.0144 | 1.91 | 68.5 |
| [MgC60]+ (hp) | 0.02 | 0.08 | 0.0203 | −0.0199 | 0.0004 | 2.45 | 31.5 |
| [CaC60]+ (p) | 0.01 | 0.04 | 0.0100 | −0.0087 | 0.0012 | 2.97 | 24.1 |
| [ScC60]+ (hh) | 0.09 | 0.15 | 0.0689 | −0.1000 | −0.0311 | 2.07 | 82.4 |
| [TiC60]+ (h) | 0.04 | 0.18 | 0.0427 | −0.0410 | 0.0017 | 2.30 | 48.8 |
| [VC60]+ (h) | 0.03 | 0.11 | 0.0327 | −0.0352 | −0.0024 | 2.36 | 42.5 |
| 0.16 | 0.0440 | −0.0468 | −0.0027 | ||||
| [CrC60]+ (hh) | 0.06 | 0.20 | 0.0617 | −0.0719 | −0.0102 | 2.20 | 40.4 |
| [MnC60]+ (h) | 0.02 | 0.06 | 0.0170 | −0.0190 | −0.0019 | 2.70 | 31.1 |
| [FeC60]+ (hh) | 0.10 | 0.17 | 0.0908 | −0.1379 | −0.0471 | 1.97 | 56.6 |
| [CoC60]+ (hh) | 0.11 | 0.16 | 0.0966 | −0.1518 | −0.0552 | 1.94 | 59.0 |
| [NiC60]+ (hh) | 0.11 | 0.15 | 0.1009 | −0.1633 | −0.0624 | 1.94 | 57.3 |
| [CuC60]+ (hh) | 0.02 | 0.15 | 0.1062 | −0.1743 | −0.0680 | 1.92 | 55.7 |
| [ZnC60]+ (p) | 0.05 | 0.16 | 0.0353 | −0.0367 | −0.0013 | 2.21 | 43.9 |
A BCP where ∇2(ρ) < 0 is dominated by a local reduction of potential energy (i.e. a covalent interaction), while a BCP where ∇2(ρ) > 0 is dominated by a local excess of kinetic energy (i.e. an ionic interaction). However, the Laplacian of the electron density value does not always give an accurate description of the interaction due to the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 kinetic:potential ratio.68 The ∇2(ρ) contour maps give a visual picture of the interaction depending on the shape of force lines: closed nodal contours are characteristic of ionic interactions while lines of forces of two different atomic nuclei mixing together are characteristic of covalent interactions.
:1 kinetic:potential ratio.68 The ∇2(ρ) contour maps give a visual picture of the interaction depending on the shape of force lines: closed nodal contours are characteristic of ionic interactions while lines of forces of two different atomic nuclei mixing together are characteristic of covalent interactions.
For alkali metal complexes we observe that the electron density is localized in spherical contours around the alkali nuclei in the manner characteristic of two separated closed-shell distributions. This points to an ion-induced dipole interaction between the two fragments: the metal cation and the polarized carbon cage. A similar behavior is observed in the alkaline earth complexes [MgC60]+ and [CaC60]+. However [BeC60]+ shows an intriguing behavior for its 2D ∇2(ρ) contour: it does not present a closed nodal shape and, furthermore, it mixes with the contours of the carbon atoms in the hp bond. Though the value of ∇2(ρ) is positive in the BCP, i.e. an ion-induced dipole interaction, the atomic basins around the interatomic surface exhibit an opposite behavior, indicating that the interaction has both covalent and ionic character: a direct ionic bond (see the bond path) and an indirect covalent character shown by the lateral density spreading in the ∇2(ρ) map. Polarization of the electronic density around the Be nucleus is also observed. This might be the reason for a kind of sp hybridization to form bonds with the lateral dispersion density of the C atoms in the hp bond, explaining the large interaction energy.
For the transition metal complexes we have found that ∇2(ρ) also exhibits closed nodal shaped contours around the metal atom, i.e. ion-induced dipole interaction. Nevertheless, we observe an increasing covalent character as we progress in the series: the electron density localized on the carbon atoms of the hh bond, points towards the metal atom (as shown in the ∇2(ρ) map), implying an increasing covalent character in the metal–fullerene interaction. Table 3 shows the values of the electron density ρ, the Laplacian of the electron density, ∇2(ρ), the kinetic energy density, G, the potential energy density, V, and the total electronic energy density, H, at the metal–fullerene BCPs for the most stable isomer of each [MC60]+ complex. The potential energy density V is always negative and gives the contribution of the covalent character to the interaction; the kinetic energy density G is always positive and provides the contribution of the ionic character. The sum of both give the total electronic energy density H, and hence one can easily predict the character of the interaction with the sign of H in the BCP. This way we can thus determine the ionic or covalent character in the metal–fullerene interaction of the studied complexes. Our results in Table 3 show that in all complexes there is very low density accumulation within the M–C60 bonds, indicating that this interaction is essentially closed-shell, thus pointing to an ion-induced dipole interaction, corroborating our previous assumptions. In the same table we can also see the previously mentioned inconsistencies between the signs of ∇2(ρ) and H. For all transition metals, the former predicts an ion-induced dipole interaction while the latter, on the contrary, predicts a covalent interaction (except for Ti). Since ρ in the BCP is small, the covalent character is not dominant, resulting an ion-induced dipole with a certain amount of covalent character.
A more quantitative analysis of the interaction between the metal cation and the fullerene has been carried out within the localized molecular orbital energy decomposition analysis (LMO-EDA).55 It allows to divide the total interaction energy (Eint) into five contributions: electrostatic, exchange, repulsion, polarization and dispersion:
| Eint = Eelec + Eexch + Erep + Epol + Edisp | (2) |
The electrostatic energy term describes the attractive or repulsive classical Coulomb interaction; the exchange term includes the Pauli repulsion; the repulsion term includes the contribution due to the exchange of electrons of parallel spin between the monomers (C60 and M+ in our case); the polarization term accounts for the energy gain by orbital relaxation of one monomer due to the presence of the undistorted charge distribution of the second monomer in its vicinity; and the dispersion contribution is the result of the instantaneous correlation of fluctuating electron density distributions.
The total interaction energy obtained within the LMO-EDA analysis (Fig. 2) correctly reproduces the metal adsorption energies obtained from all-electron calculations. Only a constant vertical displacement of ∼6 kcal mol−1 is observed. Fig. 6 shows the results of this decomposition. In all cases, but in Be, the repulsion is the only component with a positive value, i.e. it destabilizes the interaction. All the other components contribute to the stabilization of the complexes. In the same figure we can see that, for all complexes, the largest stabilizing contribution is the polarization energy. The sum of the polarization and repulsion terms leads to only negative values for the alkali complexes; for the other metals, the polarization is not enough to overcome the repulsion. In the latter systems, the exchange and the electrostatic interactions are needed to obtain positive binding energies. The dispersion energies make minor contributions to the total binding energies for all complexes. Interestingly, Fig. 6 also shows two trends in the values of the transition metals: from Sc to Mn and from Fe to Zn. In the same figure Sc shows larger contributions of each component. Fig. 7 shows the relative contribution of each component to the total stabilization of the interaction. The polarization is in all cases (except in Be) the most important stabilizing component, being dominant in the case of alkaline and alkaline-earth complexes and decreasing up to ∼50% for the transition metals. In the transition metal complexes, the electrostatic and the exchange components contribute with ∼20–30% each to stabilize the interaction. Dispersion is less than 10% in all cases.
 | ||
| Fig. 6 Energy decomposition analysis for the [MC60]+ complexes. The interaction energy is decomposed in electronic, exchange, repulsion, polarization and dispersion components (values in kcal mol−1). | ||
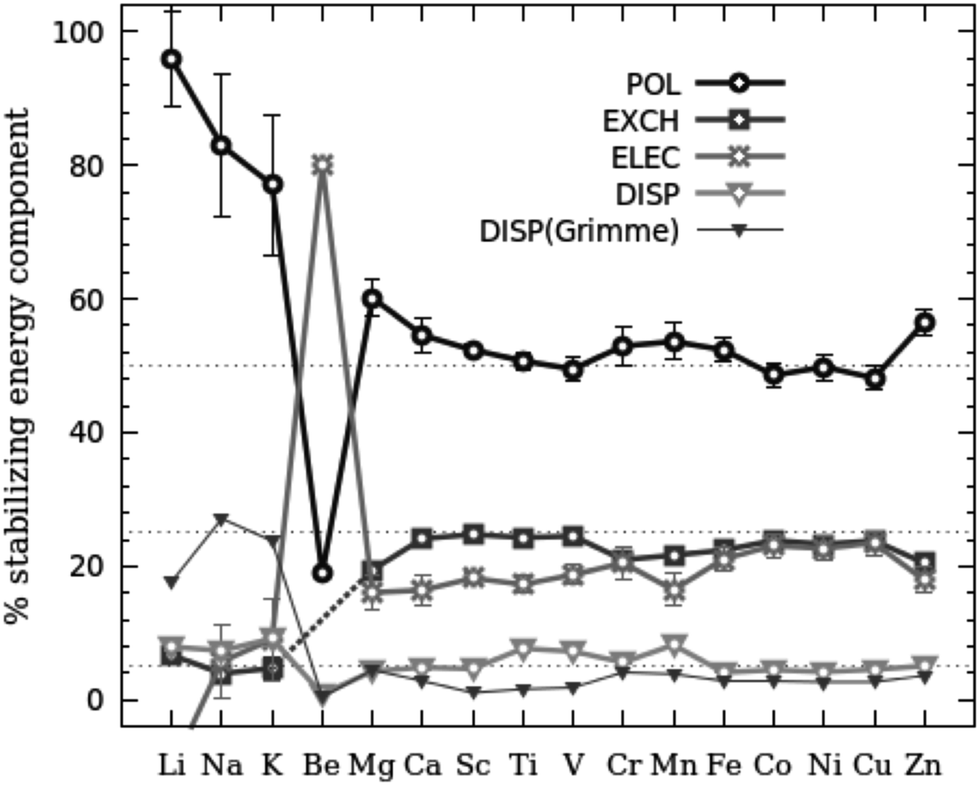 | ||
| Fig. 7 Contribution to the stabilization energy in percentage of each component in the [MC60]+ complexes. | ||
It is well known that conventional DFT functionals do not provide an appropriate description of the dispersion interactions. In practice, from a DFT perspective, dispersive interactions are a long-range correlation phenomenon that is very difficult to take into account accurately. One popular strategy is the DFT-D2 functional proposed by Grimme69 which adds an additional pairwise C6/R6 correction term to the total DFT energy in order to account for dispersion forces (EDFT-D2 = EDFT + EDISP(Grimme)). The corrections for dispersion effects calculated by using the DFT(B3LYP)-D2 scheme are shown in Fig. 7. In average, EDISP(Grimme) is −6 kcal mol−1 and the EDISP calculated by DFT(B3LYP) is −9 kcal mol−1, which indicates that the dispersion component of DFT(B3LYP) provides enough precision for this kind of systems, presumably because the dispersion component of the interaction plays a minor role at the equilibrium distances.
Thus, the energy decomposition analysis points out that the metal–fullerene interaction is of the ion-induced dipole type, as was qualitatively shown by the Bader analysis. The metal ion polarizes the π electronic cloud and the ion-induced dipole interaction dominates (Epol > 40% in all cases except in Be). In transition metal complexes other type of interaction such as Eelec and Eexch also contribute to the interaction, but in a lower extent than polarization.
Both the topological density and the energy decomposition analysis have shown that the metal fullerene interaction is dominated by an ion-induced dipole type. We propose a simple model to further rationalize these results. Based on the fact that the metal atoms in the complexes present a positive charge, and considering that the fullerene electronic cloud is easily polarizable, we assume an ion-induced dipole attraction with an interaction energy, EIiD, given by:
 | (3) |
 | (4) |
Fig. 8 shows the computed adsorption energy as a function of q2/r4, considering q = +1 and taking r the smallest metal–carbon atom distance. For most of the complexes the results follow a straight line, showing that an ion-induced dipole interaction is the dominant effect that explains the metal–fullerene bonding i.e., in these complexes the interaction between the metal atom and the fullerene is dominated by the polarization (as shown in the energy decomposition analysis). Sc appears out of the trend in this figure; as previously explained this complex shows a larger covalent character and thus, a simple ion-induced dipole model does not predict correctly the interaction energy.
 | ||
| Fig. 8 Computed adsorption energy Eads and polarization component of the interaction Epol (in kcal mol−1) as a function of an ion-induced dipole energy model, EIiD ∝ r4 (in Å−4). Dotted lines are given as guides to the eye. | ||
In a more detailed analysis of the ion-induced polarization model, we plot Epol as a function of r−4 (see also Fig. 8). In this case we can see two trends. One of them corresponds to a pure metal-ion inducing polarization of the fullerene cloud (typically for the alkali complexes). However, for a few complexes a second trend is observed for those species whose metals cations presents the largest polarizability (see Fig. 9). In these complexes the polarization of the metal electronic cloud, exerted by the dipole in the fullerene, contributes significantly to the polarization component and it is not taken into account into the simple ion-induced dipole model. Also in this figure the Sc complex lies out of the trend due to the larger covalent character.
 | ||
| Fig. 9 Polarizabilities of the cations involved in the interaction with C60, computed at the aug-pc-4/CCSD(T) level of theory (see Computational details). | ||
4 Conclusions
This work presents a systematic theoretical study of the exohedral interaction between a positively charged metal atom and a C60 fullerene. We have found that the metal atom in the [MC60]+ complexes hold the positive charge. The partial negative charge of the carbon atoms closer to the metal one reflects a localized reactivity and a strong polarization of the C60 π electronic cloud. The interaction between the metal cation and the carbon cage has been analyzed by using a simple ion-induced dipole model. This electrostatic model explains the type of interaction for most of the complexes. For those complexes that deviate from this trend, a further partial covalent character has to be taken into account. These assumptions have been confirmed qualitatively in a visual inspection of contour maps of the Laplacian of the electronic density. Furthermore, a quantitative explanation of the adsorption energy (bond strength) is given by the value of the total electronic energy density in the bond critical point. An energy decomposition analysis allows us to further confirm these findings, being the polarization component the most stabilizing one.This analysis leads us to conclude that, while the metal–fullerene interaction is dominantly ion-induced dipole in the case of alkali and alkali-earth metals (except for beryllium), it has an increasing covalent character as one moves along the first period of transition metals.
Acknowledgements
We acknowledge the generous allocation of computer time at the Centro de Computación Científica at the Universidad Autónoma de Madrid (CCC-UAM). Work partially supported by the projects FIS2010-15127, CTQ2010-17006 and CSD2007-00010 (MICINN), CTQ2013-43698-P and FIS2013-42002-R (MINECO) and S2009/MAT1726 (CAM). M.R. acknowledges that this work was partially supported by the Erasmus Mundus Program of the European Union and the FPI grant associated to the project CTQ2010-17006 of the Spanish Ministerio of Economa y Competitividad. S. D.-T. gratefully acknowledges the “Ramón y Cajal” program of the Spanish Ministerio de Educación y Ciencia.References
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- W. Kratschmer, L. D. Lamb, K. Fostiropoulos and D. R. Huffman, Nature, 1990, 347, 354–358 CrossRef.
- R. C. Haddon, A. F. Hebard, M. J. Rosseinsky, D. W. Murphy, S. J. Duclos, K. B. Lyons, B. Miller, J. M. Rosamilia, R. M. Fleming, A. R. Kortan, S. H. Glarum, A. V. Makhija, A. J. Muller, R. H. Eick, S. M. Zahurak, R. Tycko, G. Dabbagh and F. A. Thiel, Nature, 1991, 350, 320–322 CrossRef CAS.
- A. F. Hebard, M. J. Rosseinsky, R. C. Haddon, D. W. Murphy, S. H. Glarum, T. T. M. Palstra, A. P. Ramirez and A. R. Kortan, Nature, 1991, 350, 600–601 CrossRef CAS.
- M. J. Rosseinsky, A. P. Ramirez, S. H. Glarum, D. W. Murphy, R. C. Haddon, A. F. Hebard, T. T. M. Palstra, A. R. Kortan, S. M. Zahurak and A. V. Makhija, Phys. Rev. Lett., 1991, 66, 2830–2832 CrossRef CAS.
- K. Tanigaki, I. Hirosawa, T. Ebbesen, J. Mizuki and S. Kuroshima, Chem. Phys. Lett., 1993, 203, 33–36 CrossRef CAS.
- D. M. Poirier, D. W. Owens and J. H. Weaver, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 1830–1843 CrossRef CAS.
- K. Tanigaki, S. Kuroshima, J. Fujita and T. W. Ebbesen, Appl. Phys. Lett., 1993, 63, 2351–2353 CrossRef CAS PubMed.
- K. Tanigaki, I. Hirosawa, T. Ebbesen, J. Mizukishimakawa, Y. Kubo, J. Tsai and S. Kuroshima, Nature, 1992, 356, 419–421 CrossRef CAS.
- T. P. Martin, N. Malinowski, U. Zimmermann, U. Naher and H. Schaber, J. Chem. Phys., 1993, 99, 4210–4212 CrossRef CAS PubMed.
- F. Rabilloud, J. Phys. Chem. A, 2010, 114, 7241–7247 CrossRef CAS PubMed.
- J. Kohanoff, W. Andreoni and M. Parrinello, Chem. Phys. Lett., 1992, 198, 472–477 CrossRef CAS.
- J. Bretón, J. González-Platas and C. Girardet, J. Chem. Phys., 1993, 99, 4036, DOI:10.1063/1.466099.
- A. Ruiz, J. Hernández-Rojas, J. Bretón and J. M. Gómez Llorente, J. Chem. Phys., 1998, 109, 3573, DOI:10.1063/1.476953.
- U. Zimmermann, N. Malinowski, U. Näher, S. Frank and T. P. Martin, Phys. Rev. Lett., 1994, 72, 3542–3545 CrossRef CAS.
- T. Akasaka and S. Nagase, Endofullerenes: A New Family of Carbon Clusters, Kluwer Academic Publishers, 2002 Search PubMed.
- F. Tast, N. Malinowski, S. Frank, M. Heinebrodt, I. M. L. Billas and T. P. Martin, Phys. Rev. Lett., 1996, 77, 3529–3532 CrossRef CAS.
- M. Pederson, D. Porezag, D. Patton and E. Kaxiras, Chem. Phys. Lett., 1999, 303, 373–378 CrossRef CAS.
- Q. Sun, Q. Wang, P. Jena and Y. Kawazoe, J. Am. Chem. Soc., 2005, 127, 14582–14583 CrossRef CAS PubMed.
- D. Vijay, H. Sakurai, V. Subramanian and G. N. Sastry, Phys. Chem. Chem. Phys., 2012, 14, 3057–3065 RSC.
- A. V. Zabula, S. N. Spisak, A. S. Filatov, V. M. Grigoryants and M. A. Petrukhina, Chem.–Eur. J., 2012, 18, 6476–6484 CrossRef CAS PubMed.
- T. Amaya, H. Sakane and T. Hirao, Angew. Chem., Int. Ed., 2007, 46, 8376–8379 CrossRef CAS PubMed.
- M. Hirscher, M. Becher, M. Haluska, A. Quintel, V. Skakalova, Y.-M. Choi, U. Dettlaff-Weglikowska, S. Roth, I. Stepanek, P. Bernier, A. Leonhardt and J. Fink, J. Alloys Compd., 2002, 330–332, 654–658 CrossRef CAS.
- W. Huang, X. Zhang, J. Tu, F. Kong, J. Ma, F. Liu, H. Lu and C. Chen, Mater. Chem. Phys., 2003, 78, 144–148 CrossRef.
- R. Zacharia, K. Y. Kim, A. F. Kibria and K. S. Nahm, Chem. Phys. Lett., 2005, 412, 369–375 CrossRef CAS PubMed.
- M. Yoon, S. Yang, C. Hicke, E. Wang, D. Geohegan and Z. Zhang, Phys. Rev. Lett., 2008, 100, 206806 CrossRef.
- P. Jena, J. Phys. Chem. Lett., 2011, 2, 206–211 CrossRef CAS.
- C. I. Contescu, K. van Benthem, S. Li, C. S. Bonifacio, S. J. Pennycook, P. Jena and N. C. Gallego, Carbon, 2011, 49, 4050–4058 CrossRef CAS PubMed.
- Y. Guo, K. Jiang, B. Xu, Y. Xia, J. Yin and Z. Liu, J. Phys. Chem. C, 2012, 116, 13837–13841 CAS.
- Q. Sun, P. Jena, Q. Wang and M. Marquez, J. Am. Chem. Soc., 2006, 128, 9741–9745 CrossRef CAS PubMed.
- Q. Wang, Q. Sun, P. Jena and Y. Kawazoe, J. Chem. Theory Comput., 2009, 5, 374–379 CrossRef CAS.
- T. Yildirim and S. Ciraci, Phys. Rev. Lett., 2005, 94, 175501 CrossRef CAS.
- Y. Zhao, Y.-H. Kim, A. C. Dillon, M. J. Heben and S. B. Zhang, Phys. Rev. Lett., 2005, 94, 155504 CrossRef.
- O. Echt, A. Kaiser, S. Zöttl, A. Mauracher, S. Denifl and P. Scheier, ChemPlusChem, 2013, 78, 910–920 CrossRef CAS.
- C. Leidlmair, Y. Wang, P. Bartl, H. Schöbel, S. Denifl, M. Probst, M. Alcami, F. Martin, H. Zettergren, K. Hansen, O. Echt and P. Scheier, Phys. Rev. Lett., 2012, 108, 076101 CrossRef.
- B. K. Rao and P. Jena, Europhys. Lett., 1992, 20, 207–312 CrossRef.
- T. M. McCleskey and B. L. Scott, J. Occup. Environ. Hyg., 2009, 6, 751–757 CrossRef CAS PubMed.
- A. Martin-Somer, A. M. Lamsabhi, O. Mo and M. Yanez, Comput. Theor. Chem., 2012, 998, 74–79 CrossRef CAS PubMed.
- S. Metz, M. C. Holthausen and G. Frenking, Z. Anorg. Allg. Chem., 2006, 632, 814–818 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision B.01, 2010, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, USA, 1994 Search PubMed.
- T. A. Keith, AIMAll 11.10.16, TK Gristmill Software, 2011, http://aim.tkgristmill.com/ Search PubMed.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066–4073 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1 Search PubMed.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS PubMed.
- C. Sporea, F. Rabilloud and M. Aubert-Frecon, J. Mol. Struct.: THEOCHEM, 2007, 802, 85–90 CrossRef CAS PubMed.
- C. Sporea and F. Rabilloud, J. Chem. Phys., 2007, 127, 164306 CrossRef CAS PubMed.
- N. Hamamoto, J. Jitsukawa and C. Satoko, Eur. Phys. J. D, 2002, 19, 211 CAS.
- K. Hedberg, L. Hedberg, D. S. Bethune, C. A. Brown, H. D. Dorn, R. D. Johnson and M. de Vries, Science, 1991, 254, 410, DOI:10.1126/science.254.5030.410.
- P. Su and H. Li, J. Chem. Phys., 2009, 131, 014102 CrossRef PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- M. Gordon and S. M. W., Advances in electronic structure theory: GAMESS a decade later, Elsevier, 2005 Search PubMed.
- http://www.theochem.uni-stuttgart.de/pseudopotentials/.
- S. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and A. Wolf, MOLPRO, version 2010.1, a package of ab initio programs, 2010, see http://www.molpro.net Search PubMed.
- F. Jensen, J. Phys. Chem. A, 2007, 111, 11198–11204 CrossRef CAS PubMed.
- F. Jensen, J. Chem. Phys., 2012, 136, 114107 CrossRef PubMed.
- F. Jensen, J. Chem. Phys., 2013, 138, 014107 CrossRef PubMed.
- J. Roques, F. Calvo, F. Spiegelman and C. Mijoule, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 205412 CrossRef.
- M. Robledo, F. Martin, M. Alcami and S. Diaz-Tendero, Theor. Chem. Acc., 2013, 132, 1346 CrossRef PubMed.
- A. Kramida, Y. Ralchenko, J. Reader and NIST ASD Team, NIST Atomic Spectra Database (ver. 5.1), National Institute of Standards and Technology, Gaithersburg, MD, 2013, http://www.physics.nist.gov/asd, [2014 April 14] Search PubMed.
- T. Guo, G. K. Odom and G. E. Scuseria, J. Phys. Chem., 1994, 98, 7745–7747 CrossRef CAS.
- D. Cremer and E. Kraka, Angew. Chem., Int. Ed., 1984, 23, 627–628 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |