
Comparison of second-order nonlinear optical chromophores with D–π–A, D–A–π–A and D–D–π–A architectures: diverse NLO effects and interesting optical behavior
Fenggang Liuab,
Yuhui Yangab,
Shengyu Congab,
Haoran Wangab,
Maolin Zhangab,
Shuhui Bo*a,
Jialei Liua,
Zhen Zhena,
Xinhou Liua and
Ling Qiu*a
aKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, PR China. E-mail: boshuhui@mail.ipc.ac.cn; qiuling@mail.ipc.ac.cn; Fax: +86-01-2543529; Tel: +86-01-82543529
bUniversity of Chinese Academy of Sciences, Beijing 100043, PR China
First published on 3rd October 2014
Abstract
Four second-order nonlinear optical chromophores with D–π–A, D–A–π–A and D–D–π–A architectures have been synthesized and systematically characterized. Chromophores A and C have been synthesized with an additional acceptor (–CN) or donor group (thiophene) on the π bridge, termed the D–A–π–A and D–D–π–A configurations. D–π–A structural chromophores B and D were chosen as reference compounds for comparison. The results show that incorporation of the –CN group could increase poling efficiency possibly due to reduced intermolecular dipole–dipole interactions which results in comparable r33 values (48 pm V−1). An r33 value of 45 pm V−1 was obtained for the film C/APC suggesting significant site isolation. Compared with the D–π–A structural chromophores B and D, chromophores A and C demonstrated similar or enhanced NLO effects and better optical transparency.
1. Introduction
In recent years, organic electro-optic (OEO) materials have attracted much attention and been extensively studied owing to their potential applications in optical switches, optical sensors, information processors, and telecommunications etc.1–4 The second-order nonlinear optical (NLO) chromophores are the key constructing blocks for OEO materials.5 To meet the stringent requirements for the using of devices, NLO chromophores should exhibit good properties such as high thermal and chemical stabilities, large nonlinearity, and good transparency, as well as easy syntheses. However, experimentally observed trade-offs, such as nonlinearity-transparency trade-off make it hard to achieve all of these requirements simultaneously.6,7 In addition, organic NLO chromophores, especially those demonstrating high β values, typically have large dipole moments. The strong inter chromophore dipole–dipole interactions may lead to unfavorable antiparallel packing of the chromophores, thus reducing the optical nonlinearities.8 Several approaches have been found to solve these problems in some degree.6,7,9,10The second-order NLO chromophores are based on a push–pull system, which consists of an electron-donating group (donor) and an electron-withdrawing group (acceptor) coupled through a π-conjugated bridge.11,12 To date, most of these chromophores have been constructed with a typical electron donor–π bridge–electron acceptor (D–π–A) configuration. Previous studies have shown that, we could construct different types of structural motifs by inserting electron-rich ring or an electron-poor ring into the π bridge of chromophore, which may lead to enhanced strength of the donor and acceptor.13–15
Recently, it has been found that incorporation of additional electron-withdrawing units (such as cyanovinyl, thiazole) into the π bridge as internal acceptors, termed the “D–A–π–A” configuration, displays several advantages such as tuning of the molecular energy levels, red-shift of the charge-transfer absorption band. They had been widely studied in the dye-sensitized solar cells (DSCs) area.16–18 There were articles about tricyanopyrrolidene-based chromophores containing thiophene as a lateral moiety to the π bridge which form the “D–D–π–A” configuration.19,20 Based on the excellent work on the different structures of dyes, we were wondering if it is possible to introduce additional acceptor or donor group on the π bridge but without changing the conjugated length of the chromophores. Perhaps, the introduced acceptor or donor group could isolate the chromophores from each other more effectively, thus improving the NLO effect. And they may have different influences on electron transfer and optical properties.
So, in our paper, we would like to compare the structure–property relationship between the three architectures of the second-order chromophores: D–π–A, D–A–π–A and D–D–π–A configurations. We had designed and synthesized chromophores A–D based on the 4-(diethylamino) aryl group donor and strong tricyanovinyldihydrofuran (TCF) acceptor in simple three or four step reactions. Chromophores A and C had been synthesized with additional acceptor (–CN) or donor group (thiophene) on the π bridge to form the D–A–π–A and D–D–π–A configurations. D–π–A structural chromophores B and D were chosen as reference compounds for comparison. The synthesis, UV-vis, solvatochromic, DFT calculations, thermal stabilizes and EO activities of these chromophores were systematically studied and compared to illustrate the architectural influence on rational NLO chromophore designs (Chart 1).
 | ||
| Chart 1 Chemical structure for chromophores A–D. | ||
2. Results and discussion
2.1 Synthesis and characterization of chromophores
Scheme 1 shows the synthetic approach for the new chromophores A–D. Starting from the amine donor aldehyde intermediate compound 3, chromophores A–D were synthesized in good overall yields through simple three or four step reactions: the free hydroxyl group on the donor was protected by the alkyl group to improve the solubility through Williamson ether synthesis, after introduction of the bridge by Wittig condensation, compounds 4a–c were prepared with a high yield. Treatment of compound 4b with POCl3 and DMF gave an aldehyde 5c. Treatment of compound 4a–b with n-BuLi and DMF gave an aldehyde 5a–b. And the final condensations with the TCF acceptor give chromophores A and B as green solids and chromophores C and D as blue solids. All of the chromophores were fully characterized by 1H-NMR, 13C-NMR, elemental analysis and mass spectroscopy. These chromophores possess good solubility in common organic solvents, such as dichloromethane, chloromethane and acetone. | ||
| Scheme 1 Chemical structures and synthetic routes for chromophores A–D. | ||
2.2 Thermal stability
The NLO chromophores must be thermally stable in order to withstand the poling process and subsequent processing for use. The thermal stability of the chromophores was investigated using thermo gravimetric analysis (TGA) as shown in Fig. 1. All the chromophores exhibited good thermal stabilities with the decomposition temperatures (Td) higher than 200 °C as showed in Table 1. The chromophore D had the minimum decomposition temperature (Td = 200 °C). It may be attributed to the reason that the extended polyenes are typically not sufficiently stable.21 The chromophore A–C showed different decomposition temperatures from 225 °C to 236 °C, which was high enough for the application in EO device preparation.22 | ||
| Fig. 1 TGA curves of chromophores A–D with a heating rate of 10 °C min−1 in nitrogen atmosphere. | ||
2.3 Optical properties
In order to reveal the effect of different structures on the intramolecular charge transfer (ICT) of dipolar chromophores, UV-vis absorption spectra of the four chromophores were measured in series solvents with different dielectric constants, as shown in Fig. 2.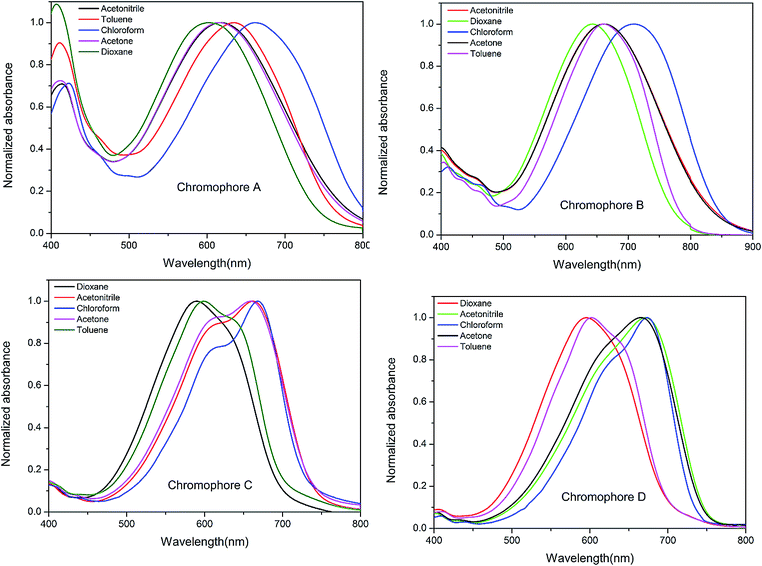 | ||
| Fig. 2 UV-vis absorption spectra of chromophore A–D in six kinds of aprotic solvents with varying dielectric constants (ε). | ||
The spectrum data are summarized in Table 1. Depending on different structures, the resulting NLO chromophores exhibited diverse intramolecular charge-transfer (ICT) absorption properties. It may see from the Fig. 2 that chromophore A, B, C and D showed the maximum absorption (λmax) from 663 nm to 709 nm in chloroform. Incorporation of additional electron-withdrawing –CN unit into the π bridge did not red-shift the maximum absorption band as reported in the dye-sensitized solar cells (DSCs) molecules containing cyanoacrylic acid acceptor.16–18 Compared to chromophore B, chromophore A showed large blue-shift (Δλ = 46 nm), it attribute to the different acceptors: the –CN group would enhance the electron-withdrawing ability of cyanoacrylic acid, but such an enhancement may weaken if the acceptor become much stronger acceptor TCF. And, it may somehow weak the strength of the donor. Chromophores C and D exhibit the absorption maximum (λmax) at 668 and 673 nm in chloroform, respectively. This may be explained by that chromophore C possessed a less coplanar geometry due to the steric hindrance of the lateral group thiophene.
Besides, the solvatochromic behavior was also an important aspect to investigate the polarity of chromophores. It was found that both chromophores C and D showed very large bathochromic shifts of 79 and 78 nm, from dioxane to chloroform, displaying larger solvatochromism than CLD (60 nm) and the similar single donor FTC (61 nm).23,24 Chromophore A and B showed moderate bathochromic shifts of 61 and 66 nm from dioxane to chloroform. This confirms that chromophores C and D are more easily polarizable than chromophore A and B.9,25 With a further increase of solvent polarity, a saturation behavior was found for four chromophores in more polar solvents, such as acetone, and acetonitrile. For example, compounds A and B showed hypsochromic shifts of −45 nm and −47 nm, respectively, from chloroform to acetone. Chromophores C and D also exhibited similar solvatochromic behavior. Such a phenomenon was reported by Davies9 and was ascribed to the back electron transfer from the acceptor side to the donor side in polar solvents which causing blue-shift from the absorption.14
2.4 Theoretical calculations
The DFT calculations were carried out at the hybrid B3LYP level by employing the split valence 6-311g (d, p) basis set26,27 to understand the ground-state polarization of the chromophores with different configurations. All molecules were assumed to be in trans-configurations. The HOMO–LUMO energy gap, dipole moment (μ), and first hyperpolarizability (β) of the chromophores obtained from DFT calculations are summarized in Table 2.| Chromophore | ΔE (DFT)a (V) | HOMOb (eV) | LUMOc (eV) | βtotd (10−30 esu) | μe (D) |
|---|---|---|---|---|---|
a ΔE (DFT) was calculated from DFT calculations.b calculated from DFT calculations.c calculated from DFT calculations.d βtot is the first-order hyperpolarizability, and where  , calculated from DFT quantum mechanical methods.e μ is the total dipole moment. , calculated from DFT quantum mechanical methods.e μ is the total dipole moment. |
|||||
| A | 2.12 | −5.580 | −3.459 | 518.57 | 15.05 |
| B | 2.03 | −5.290 | −3.259 | 678.72 | 20.53 |
| C | 2.49 | −5.514 | −3.018 | 208.61 | 20.67 |
| D | 2.44 | −5.507 | −3.065 | 226.73 | 21.49 |
The HOMO–LUMO energy gap was used to understand the charge transfer interaction occurring in a chromophore molecule.22,28 Fig. 3 depicts the electron density distribution of the HOMO and LUMO structures. It can be seen that the density of the ground and excited state electron is asymmetry along the dipolar axis of the chromophores.
 | ||
| Fig. 3 The frontier molecular orbitals of chromophores A–D. | ||
The HOMO and LUMO energies were calculated by DFT method and summarized in Table 2. As showed in table, the HOMO energy of chromophore A is 0.29 eV lower than that of chromophore B (−5.580 eV vs. −5.290 eV), while the LUMO energy of chromophore A is 0.2 eV lower than that of chromophore B (−3.459 eV vs. −3.259 eV). The cyanovinyl unit has the potential of lowering the LUMO energy and hence a lower excitation energy.29 However, in chromophore A, the –CN unit also had potential of lowering the HOMO energy to a greater extent than the LUMO energy. These two factors increase the gap between the HOMO and LUMO by 0.09 eV. The HOMO–LUMO gap of chromophore C was little larger than chromophore D, this may be explained by that conjugate structure was somehow wrecked by the lateral group thiophene. As ΔE is reduced, resulting in a bathochromic shift of λmax within the series of compounds. These results correspond with the conclusion of UV-vis spectra analysis.
To get more information from the frontier orbitals, the composition of the HOMOs and LUMOs has been calculated using the Multiwfn program.30 As shown in Table 3, the whole chromophore molecule was segmented as donor, π-bridge, and acceptor. At the same time, the attributions of the substituent groups (–CN, thiophene) located in bridge moiety were listed separately. For the chromophores A and B, the LUMO was largely stabilized by the contributions from acceptors (43.76–44.27%) and the π-bridges (38.99–39.77%), while the HOMO was largely stabilized by the contributions from donors (46.57–52.21%) and π-bridges (28.95–30.93%). For the chromophores C and D, the LUMO and HOMO were largely stabilized by the contributions from acceptors and donors. The difference attribute to the different properties of thiophene and polyenes. For chromophore C, when compare LUMO to HOMO level, the contribution of thiophene ring decreased to 0.26% from 2.52%. The comparison of HOMO and LUMO electron distribution in the thiophene ring indicated the easy delocalization of electrons in the thiophene ring. Consequently, the thiophene ring can be treated as another donor, which efficiently enhances the electron density of the conjugated system and increases the polarizability of chromophore C.31 This corresponded well with the results of solvatochromism study. Similarly, the comparison of HOMO and LUMO electron distribution in the –CN group indicated that the –CN can be treated as another acceptor.
| Chromophore | A | B | C | D | ||||
|---|---|---|---|---|---|---|---|---|
| HOMO | LUMO | HOMO | LUMO | HOMO | LUMO | HOMO | LUMO | |
| a The molecular orbital composition was calculated using Multiwfn program with Ros-Schuit (SCPA) partition. | ||||||||
| Donor | 46.57% | 17.13% | 52.21% | 15.96% | 47.57% | 36.10% | 49.47% | 36.28% |
| π Bridge | 30.93% | 38.99% | 28.95% | 39.77% | 11.45% | 16.26% | 10.42% | 16.36% |
| Acceptor | 19.55% | 43.76% | 18.84% | 44.27% | 38.47% | 47.38% | 40.11% | 47.36% |
| Thiophene | 2.52% | 0.26% | ||||||
| CN | 2.96% | 0.13% | ||||||
First hyperpolarizability is a third rank tensor that can be described by a 3 × 3 × 3 matrix. The components of β can be calculated using the following equation:32


When used carefully and consistently, this method of DFT has been shown to give relatively consistent descriptions of first-order hyperpolarizability for a number of similar chromophores.27,33
As reported earlier, the β value has a close relationship with the substituents, steric hindrance, and intramolecular charge-transfer, π-conjugation length and so on.34,35 The β value of chromophore B was larger than that of chromophore A due to narrower energy gap between HOMO and LUMO as shown in Table 2. It attribute to the introduction of electron-withdrawing units –CN. It may weak the strength of the donor probable due to its nearby to the donor. The β value of chromophore C was little smaller than that of chromophore D. This may be explained by that chromophore C possessed a less coplanar geometry due to the steric hindrance of the lateral group thiophene.
The calculated results indicated that the total dipole moment (μ) of chromophore A is smaller than that of chromophores B–D. The –CN substitution would create another local dipole moment in other direction which may reduce the total dipole moment. The smaller μ can reduce the aggregation of chromophore A in organic materials which was in accordance with the electro-optic performance study.36
2.5 Electro-optic performance
In order to investigate the translation of the microscopic hyperpolarizability into macroscopic EO response, the polymer films doped with 25 wt% chromophores into amorphous polycarbonate (APC) were prepared using dibromomethane as solvent. The resulting solutions were filtered through a 0.2 μm PTFE filter and spin-coated onto indium tin oxide (ITO) glass substrates. Films of doped polymers were baked at 80 °C in a vacuum oven overnight. The corona poling process was carried out at a temperature of 10 °C above the glass transition temperature (Tg) of the polymer. The r33 values of poled films were measured by Teng–Man simple reflection method at a wavelength of 1310 nm using carefully selected thin ITO electrode with low reflectivity and good transparency in order to minimize the contribution from multiple reflections.37–39The measured r33 values depend on the chromophore number density (N), β value, and poling efficiency, described by the cos3(θ) order parameter, as indicated by40
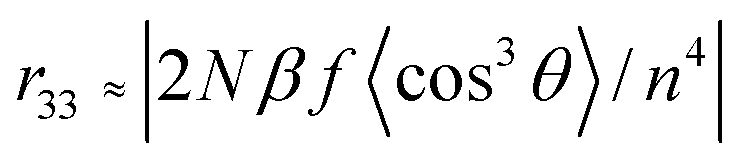
Where the f term describes electric-field factors and n is the refractive index of the film, both of which remain relatively constant for related chromophores at similar loading densities. cos3(θ) is the acentric order parameter. θ is the angle between the permanent dipole moment of chromophores and the applied electric field. At low concentration, the electro-optic activity increased with chromophore density, dipole moment and the strength of electric poling field. However, when the concentrations of chromophores increased to a certain extent, the N and cos3(θ) are no longer independent factor. Then,
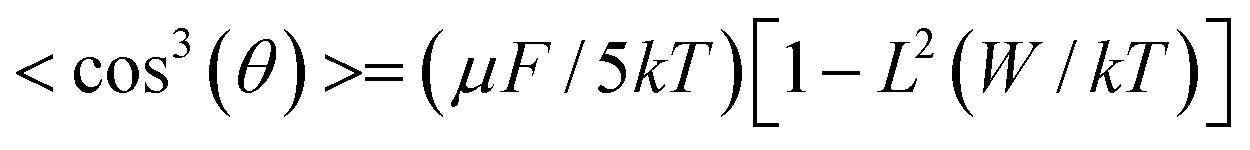
When the intermolecular electrostatic interactions are neglected, the electro-optic coefficient (r33) should increase linearly with chromophore density, first hyperpolarizability and the strength of electric poling field. But chromophores with large dipole moment generate intermolecular static electric field dipole–dipole interaction, which leads to the unfavorable antiparallel packing of chromophores. So the number of truly oriented chromophore (N) is small. In molecular optimization, introducing the huge steric hindrance group to isolate chromophores is the most popular and easy way to attenuate the dipole-dipole interactions of chromophores.41
To evaluate their EO activity, 25 wt% of the chromophores A–D were doped into APC and formulated as the typical guest–host polymer. For this series of chromophores, the poled films of A/APC, B/APC, C/APC and D/APC afforded r33 values of 48, 52, 45 and 21 pm V−1, respectively. In order to conclusively determine the relative contributions of β values to the r33 values obtained for the four chromophore with quite different β value, further investigation is necessary.
To identify the underlying mechanism for the difference in achievable EO performance, the order parameter (Φ) was calculated by measuring UV-vis absorption spectra. After the corona poling, the dipole moments of the chromophore moieties in the polymer were aligned, and the absorption curve decreased due to birefringence.25 The order parameter (Φ) for films can be calculated from the absorption changes according to the following equation: Φ = 1 − A/A0,42 in which A and A0 are the respective absorptions of the polymer films after and before corona poling. The order parameter (Φ) of poled films was calculated. The Φ values of film-A/APC, B/APC, C/APC and D/APC are 22.48% 16.05%, 28.95%, 13.52% respectively as shown in Fig. 4. This suggests the poling efficiencies of the four chromophores may indeed be quite different.43
 | ||
| Fig. 4 UV-vis absorption spectra of E-O polymers before and after poling (25 wt%). | ||
For chromophores A and B, DFT calculations suggest that the β value of chromophore B is 30% larger than that of chromophores A, while their r33 values were nearly. The larger order parameter (22.48% and 16.05%) indicates that film-A/APC has weaker inter chromophore electrostatic interactions than film-B/APC in this density. While the small structural differences between chromophores A and chromophores B likely have little effect on the steric of poling, it may have a larger influence on the electrostatics of it. For chromophore A, the introduction of the –CN group on the π bridge produces a decrease of the dipole moment of the push–pull molecule from about 20 to 15 D, the decrease of dipole moment can efficiently reduce the intermolecular electrostatic interactions. Moreover, the side –CN group may somehow act as the isolation group to suppress the possible charge transport between chromophores. So, the film A/APC achieves similar r33 values with that of film B/APC (48 and 52 pm V−1), even though it has relatively lower β. Meanwhile, it offers a pleasant surprise to the nonlinearity-transparency trade off that almost inevitably occurs in the dipolar donor–π–acceptor systems with a 41 nm hypsochromic shift of absorption maximum.
The poled films of C/APC and D/APC afforded r33 values of 45 and 21 pm V−1, respectively, although chromophores C and chromophores D have similar β value. The EO coefficient measurements clearly indicate that the chromophores C show significant improvements in r33 value as compared to the chromophores D. The largest order parameter (28.95%) indicates that film-C/APC has weaker inter chromophore electrostatic interactions than other films in this density and chromophores C is more polarizable.43 This corresponded well with the results of solvatochromism study. When at a low-density range, the intermolecular dipolar interactions are relatively weak. The intermolecular dipole–dipole interactions would become stronger and stronger, accompanying with the increased concentration of NLO chromophore moieties in the polymer which would finally lead to a decreased NLO coefficient. As for chromophore C, it may be revealed by the optimized configurations (Fig. 5) that the side thiophene ring group was perpendicular to the direction of the dipole moment of the chromophore which could act as the isolation group to suppress the possible aggregation. The larger r33 values indicates that film-C/APC has weaker inter chromophore electrostatic interactions than film-D/APC in this density. Larger r33 values are obtained for the chromophore C than that of chromophore D (45 and 21 pm V−1), suggesting the side thiophene ring group can reduce the inter chromophore dipole–dipole interactions more effectively, thereby enhancing the NLO effects of the chromophores.
 | ||
| Fig. 5 The optimized structure of chromophore A–D. | ||
3. Conclusion
Four organic NLO chromophores based on the 4-(diethylamino) aryl group donor and strong tricyanovinyldihydrofuran (TCF) acceptor with D–π–A, D–A–π–A and D–D–π–A architectures had been synthesized and systematically characterized. The chromophores obtained high thermal and chemical stabilities, large nonlinear optical coefficients, and good solubility in all common solvents. Chromophores A and C had been synthesized with additional acceptor (–CN) or donor group (thiophene) on the π bridge. D–π–A structural chromophores B and D were chosen as reference compounds for comparison.The results show that incorporation of the –CN group could reduce intermolecular dipole–dipole interactions due to reduced μ which results in comparable r33 values (48 pm V−1). r33 values of 45 pm V−1 are obtained for the film C/APC suggesting significant site isolation. Compare with the D–π–A structural chromophores B and D, chromophores A and C demonstrated similar or enhanced NLO effects and better optical transparency. These investigations suggest that substituent groups play a critical role in affecting the nonlinearity of D–π–A chromophores, which could provide a general tool to guide the future molecular design of highly efficient nonlinear optical chromophores.
4. Experimental section
4.1 Materials and instrument
All chemicals are commercially available and were used without further purification unless otherwise stated. N,N-dimethyl formamide (DMF), phosphorusoxychloride (POCl3), tetrahydrofuran (THF) and ether was distilled over calcium hydride and stored over molecular sieves (pore size 3 Å). The 2-dicyanomethylene-3-cyano-4-methyl-2,5-dihydrofuran (TCF) acceptor was prepared according to the literature.44 TLC analyses were carried out on 0.25 mm thick precoated silica plates and spots were visualized under UV light. Chromatography on silica gel was carried out on Kieselgel (200–300 mesh).4.2 Measurements and instrumentation
1H-NMR and 13C-NMR spectra were determined by an Advance Bruker 400M (400 MHz) NMR spectrometer (tetramethylsilane as internalreference). The MS spectra were obtained on MALDI-TOF (Matrix Assisted Laser Desorption/Ionization of Flight) on BIFLEXIII (Broker Inc.) spectrometer. The UV-vis spectra were performed on Cary 5000 photo spectrometer. The TGA was determined by TA5000-2950TGA (TA co) with a heating rate of 10 °C min−1 under the protection of nitrogen. The elemental analysis was measured on Flash EA 1112 Elemental Analyzer.4.3 Synthesis and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane = 1:10) to give 4 as an orange oil in 91.3% yield (15.19 g, 54.78 mmol). 1H NMR (400 MHz, CDCl3) δ 10.18 (s, 1H, CHO), 7.70 (d, J = 9.0 Hz, 1H, ArH), 6.25 (d, J = 9.0 Hz, 1H, ArH), 6.01 (s, 1H, ArH), 4.02 (t, J = 6.3 Hz, 2H, CH), 3.41 (q, J = 7.1 Hz, 4H, CH2), 1.87–1.75 (m, 2H, CH2), 1.55–1.43 (m, 2H, CH2), 1.35 (dt, J = 7.3, 4.8 Hz, 4H, CH2), 1.21 (t, J = 7.1 Hz, 6H, CH3), 0.90 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 186.61, 163.65, 153.65, 129.72, 114.20, 104.12, 92.73, 67.91, 44.68, 31.49, 29.06, 25.75, 22.45, 13.80, 12.13. MS (EI) (M+, C17H27NO2): calcd: 277.40; found: 277.20.:hexane = 1:20) to give 4a as an orange powder in 76.5% yield (2.92 g, 7.65 mmol). 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 9.0 Hz, 1H, ArH), 7.76 (s, 1H, CH), 7.15 (m, 1H, CH), 7.10 (d, J = 5.1 Hz, 1H, CH), 6.97–6.92 (m, 1H, CH), 6.28 (m, 1H, ArH), 6.01 (d, J = 17.5 Hz, 1H, ArH), 3.93 (t, J = 6.4 Hz, 2H, CH2), 3.34 (q, J = 7.1 Hz, 4H, CH2), 1.82–1.69 (m, 2H, CH2), 1.50–1.41 (m, 2H, CH2), 1.32–1.27 (m, 4H, CH2), 1.15 (t, J = 7.1 Hz, 6H, CH3), 0.85 (q, J = 8.8 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 158.91, 140.62, 133.85, 128.00, 126.76, 123.36, 122.92, 117.93, 103.51, 93.42, 69.54, 67.25, 43.66, 30.57, 24.73, 21.68, 13.02, 11.67. MS (MALDI) (M+, C23H30N2OS): calcd: 382.56; found: 382.21.:hexane = 1:20) to give compound 4b as orange oil in 83.7% yield (1.50 g, 4.18 mmol). 1H NMR (400 MHz, CDCl3) δ 7.26 (d, J = 8.7 Hz, 1H, CH), 7.15 (d, J = 11.7 Hz, 1H, CH), 7.07 (d, J = 11.7 Hz, 1H, CH), 7.00 (d, J = 4.6 Hz, 1H, CH), 6.87 (m, 2H, CH, PhH), 6.20 (d, J = 10.3 Hz, 1H, CH), 6.09 (s, 1H, PhH), 3.92 (t, J = 6.4 Hz, 2H, CH2), 3.28 (q, J = 7.0 Hz, 4H, CH2), 1.83–1.72 (m, 2H, CH2), 1.46 (m, 2H, CH2), 1.30 (m, 4H, CH2), 1.10 (t, J = 7.0 Hz, 6H, CH3), 0.85 (t, J = 6.7 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 157.00, 147.66, 144.05, 126.80, 126.24, 123.34, 122.66, 121.42, 116.25, 112.77, 103.20, 94.93, 67.17, 43.49, 30.60, 28.37, 24.9, 21.58, 13.01, 11.72 (MALDI) (M+, C22H31NOS): calcd: 357.55; found: 357.86.:hexane = 1:10) to give compound 5a as a red solid in 74.3% yield (1.59 g, 3.89 mmol). 1H NMR (400 MHz, CDCl3) δ 9.73 (s, 1H, CHO), 8.21 (d, J = 9.2 Hz, 1H, ArH), 7.95 (s, 1H, CH), 7.57 (d, J = 4.0 Hz, 1H, CH), 7.18 (d, J = 4.0 Hz, 1H, CH), 6.26 (dd, J = 9.2, 2.4 Hz, 1H, ArH), 5.97 (d, J = 2.4 Hz, 1H, ArH), 3.92 (t, J = 6.5 Hz, 2H, CH2), 3.34 (q, J = 7.1 Hz, 4H, CH2), 1.83–1.72 (m, 2H, CH2), 1.50–1.39 (m, 2H, CH2), 1.35–1.26 (m, 4H, CH2), 1.14 (t, J = 7.1 Hz, 6H, CH3), 0.84 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 182.23, 160.62, 152.18, 140.74, 137.67, 137.52, 129.65, 124.36, 118.28, 110.05, 104.90, 95.38, 93.87, 68.46, 44.82, 31.56, 29.14, 25.88, 22.66, 14.05, 12.67 (MALDI) (M+, C24H30N2O2S): calcd: 410.57; found: 410.20.:hexane = 1:5) to give compound 5b as an orange oil in 77.1% yield (0.30 g, 0.77 mmol). 1H NMR (400 MHz, CDCl3) δ 9.69 (s, 1H, CHO), 7.46 (d, J = 4.0 Hz, 1H, CH), 7.23–7.12 (m, 1H, ArH), 7.00 (d, J = 4.0 Hz, 1H, CH), 6.70 (d, J = 11.9 Hz, 1H, CH), 6.42 (d, J = 11.9 Hz, 1H, CH), 6.13 (dd, J = 8.6, 2.3 Hz, 1H, ArH), 6.08 (d, J = 2.3 Hz, 1H, ArH), 3.87 (t, J = 6.5 Hz, 2H, CH2), 3.30 (q, J = 7.1 Hz, 4H, CH2), 1.72–1.54 (m, 2H, CH2), 1.37–1.29 (m, 2H, CH2), 1.26–1.20 (m, 4H, CH2), 1.12 (t, J = 7.1 Hz, 6H, CH3), 0.80 (t, J = 6.8 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 181.71, 157.27, 150.76, 148.69, 139.96, 135.30, 129.61, 126.94, 118.03, 111.11, 102.65, 94.32, 67.16, 43.45, 30.44, 28.27, 24.72, 21.62, 13.03, 11.70 (MALDI) (M+, C23H31NO2S): calcd: 385.56; found: 385.74.:hexane = 1:10) to give compound 5c as an orange-red solid in 73.3% yield (0.56 g, 1.47 mmol). 1H NMR (400 MHz, CDCl3) δ 9.87 (s, 0.11H, CHO), 9.52 (s, 0.89H, CHO), 8.17 (s, 0.11H, CH), 7.79 (s, 0.89H, CH), 7.31 (d, J = 6.2 Hz, 1H, CH), 7.00 (dd, J = 5.1, 3.5 Hz, 1H, CH),6.98 (m, 1H, CH) 6.95 (dd, J = 8.6, 5.1 Hz, 1H, PrH), 5.98 (d, J = 2.0 Hz, 1H, PrH), 5.95 (m, 1H, PrH), 3.95 (t, J = 6.4 Hz, 2H, CH2), 3.27 (q, J = 7.1 Hz, 4H, CH2), 1.83–1.73 (m, 2H, CH2), 1.50–1.40 (m, 4H, CH2), 1.35–1.25 (m, 2H, CH2), 1.14 (t, J = 7.1 Hz, 0.66H, CH3), 1.09 (t, J = 7.1 Hz, 5.34H, CH3), 0.84 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 191.95, 159.53, 150.49, 146.18, 134.43, 130.66, 126.45, 126.20, 125.36, 109.68, 103.06, 93.01, 67.40, 43.38, 30.47, 28.16, 24.73, 21.46, 13.00, 11.66 (MALDI) (M+, C23H31NO2S): calcd: 385.56; found: 385.80.:hexane = 1:10) to give compound 4c as orange oil in 86.7% yield (0.47 g, 1.56 mmol). 1H NMR (400 MHz, CDCl3) δ 9.55 (d, J = 8.1 Hz, 1H, CHO), 7.70 (d, J = 15.4 Hz, 1H, CH), 7.39 (d, J = 8.4 Hz, 1H, PhH), 6.60 (dd, J = 15.4, 8.1 Hz, 1H, CH), 6.28 (d, J = 8.4 Hz, 1H, PhH), 6.09 (s, 1H, PhH), 4.01 (t, J = 5.2 Hz, 2H, CH2), 3.43 (q, J = 7.1 Hz, 4H, CH2), 1.92–1.77 (m, 2H, CH2), 1.49 (m, 4H, CH2), 1.40–1.32 (m, 2H, CH2), 1.21 (t, J = 7.1 Hz, 6H, CH3), 0.93–0.89 (t, J = 7.1 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 193.31, 159.18, 150.81, 148.30, 129.80, 122.68, 103.68, 93.72, 67.39, 43.71, 30.56, 28.19, 24.84, 21.55, 12.94, 11.69. MS (EI) (M+, C19H29NO2): calcd: 303.44; found: 303.23.:hexane = 1:5) to give chromophore A as a green solid in 46.7% yield (0.14 g, 0.23 mmol). 1H NMR (400 MHz, acetone) δ 8.26 (d, J = 9.2 Hz, 1H, PrH), 8.15 (d, J = 16.0 Hz, 1H, CH), 8.06 (s, 1H, CH), 7.74 (d, J = 4.1 Hz, 1H, CH), 7.30 (d, J = 4.1 Hz, 1H, CH), 6.88 (d, J = 16.0 Hz, 1H, CH), 6.53 (dd, J = 9.2, 2.3 Hz, 1H, PrH), 6.33 (d, J = 2.3 Hz, 1H, PrH), 4.17 (t, J = 6.2 Hz, 2H, CH2), 3.57 (q, J = 7.1 Hz, 4H, CH2), 1.90 (s, 6H, CH3), 1.64–1.52 (m, 2H, CH2), 1.47–1.36 (m, 4H, CH2), 1.34–1.27 (m, 2H, CH3), 0.92 (t, J = 7.1 Hz, 3H, CH3). 13C NMR (101 MHz, acetone) δ 178.18, 175.67, 161.74, 153.72, 151.07, 140.05, 139.19, 138.16, 137.63, 130.15, 126.30, 118.91, 114.04, 112.67, 111.68, 110.89, 106.30, 98.75, 95.21, 69.26, 45.28, 32.25, 26.94, 26.21, 24.10, 23.34, 14.37, 13.01 (MALDI) (M+, C35H37N5O2S): calcd: 591.77; found: 591.23. Anal. calcd (%) for C35H37N5O2S: C, 71.04; H, 6.30; N, 11.83; found: C, 71.09; H, 6.37; N, 11.79.
:hexane = 1:10) to give 4 as an orange oil in 91.3% yield (15.19 g, 54.78 mmol). 1H NMR (400 MHz, CDCl3) δ 10.18 (s, 1H, CHO), 7.70 (d, J = 9.0 Hz, 1H, ArH), 6.25 (d, J = 9.0 Hz, 1H, ArH), 6.01 (s, 1H, ArH), 4.02 (t, J = 6.3 Hz, 2H, CH), 3.41 (q, J = 7.1 Hz, 4H, CH2), 1.87–1.75 (m, 2H, CH2), 1.55–1.43 (m, 2H, CH2), 1.35 (dt, J = 7.3, 4.8 Hz, 4H, CH2), 1.21 (t, J = 7.1 Hz, 6H, CH3), 0.90 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 186.61, 163.65, 153.65, 129.72, 114.20, 104.12, 92.73, 67.91, 44.68, 31.49, 29.06, 25.75, 22.45, 13.80, 12.13. MS (EI) (M+, C17H27NO2): calcd: 277.40; found: 277.20.:hexane = 1:20) to give 4a as an orange powder in 76.5% yield (2.92 g, 7.65 mmol). 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 9.0 Hz, 1H, ArH), 7.76 (s, 1H, CH), 7.15 (m, 1H, CH), 7.10 (d, J = 5.1 Hz, 1H, CH), 6.97–6.92 (m, 1H, CH), 6.28 (m, 1H, ArH), 6.01 (d, J = 17.5 Hz, 1H, ArH), 3.93 (t, J = 6.4 Hz, 2H, CH2), 3.34 (q, J = 7.1 Hz, 4H, CH2), 1.82–1.69 (m, 2H, CH2), 1.50–1.41 (m, 2H, CH2), 1.32–1.27 (m, 4H, CH2), 1.15 (t, J = 7.1 Hz, 6H, CH3), 0.85 (q, J = 8.8 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 158.91, 140.62, 133.85, 128.00, 126.76, 123.36, 122.92, 117.93, 103.51, 93.42, 69.54, 67.25, 43.66, 30.57, 24.73, 21.68, 13.02, 11.67. MS (MALDI) (M+, C23H30N2OS): calcd: 382.56; found: 382.21.:hexane = 1:20) to give compound 4b as orange oil in 83.7% yield (1.50 g, 4.18 mmol). 1H NMR (400 MHz, CDCl3) δ 7.26 (d, J = 8.7 Hz, 1H, CH), 7.15 (d, J = 11.7 Hz, 1H, CH), 7.07 (d, J = 11.7 Hz, 1H, CH), 7.00 (d, J = 4.6 Hz, 1H, CH), 6.87 (m, 2H, CH, PhH), 6.20 (d, J = 10.3 Hz, 1H, CH), 6.09 (s, 1H, PhH), 3.92 (t, J = 6.4 Hz, 2H, CH2), 3.28 (q, J = 7.0 Hz, 4H, CH2), 1.83–1.72 (m, 2H, CH2), 1.46 (m, 2H, CH2), 1.30 (m, 4H, CH2), 1.10 (t, J = 7.0 Hz, 6H, CH3), 0.85 (t, J = 6.7 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 157.00, 147.66, 144.05, 126.80, 126.24, 123.34, 122.66, 121.42, 116.25, 112.77, 103.20, 94.93, 67.17, 43.49, 30.60, 28.37, 24.9, 21.58, 13.01, 11.72 (MALDI) (M+, C22H31NOS): calcd: 357.55; found: 357.86.:hexane = 1:10) to give compound 5a as a red solid in 74.3% yield (1.59 g, 3.89 mmol). 1H NMR (400 MHz, CDCl3) δ 9.73 (s, 1H, CHO), 8.21 (d, J = 9.2 Hz, 1H, ArH), 7.95 (s, 1H, CH), 7.57 (d, J = 4.0 Hz, 1H, CH), 7.18 (d, J = 4.0 Hz, 1H, CH), 6.26 (dd, J = 9.2, 2.4 Hz, 1H, ArH), 5.97 (d, J = 2.4 Hz, 1H, ArH), 3.92 (t, J = 6.5 Hz, 2H, CH2), 3.34 (q, J = 7.1 Hz, 4H, CH2), 1.83–1.72 (m, 2H, CH2), 1.50–1.39 (m, 2H, CH2), 1.35–1.26 (m, 4H, CH2), 1.14 (t, J = 7.1 Hz, 6H, CH3), 0.84 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 182.23, 160.62, 152.18, 140.74, 137.67, 137.52, 129.65, 124.36, 118.28, 110.05, 104.90, 95.38, 93.87, 68.46, 44.82, 31.56, 29.14, 25.88, 22.66, 14.05, 12.67 (MALDI) (M+, C24H30N2O2S): calcd: 410.57; found: 410.20.:hexane = 1:5) to give compound 5b as an orange oil in 77.1% yield (0.30 g, 0.77 mmol). 1H NMR (400 MHz, CDCl3) δ 9.69 (s, 1H, CHO), 7.46 (d, J = 4.0 Hz, 1H, CH), 7.23–7.12 (m, 1H, ArH), 7.00 (d, J = 4.0 Hz, 1H, CH), 6.70 (d, J = 11.9 Hz, 1H, CH), 6.42 (d, J = 11.9 Hz, 1H, CH), 6.13 (dd, J = 8.6, 2.3 Hz, 1H, ArH), 6.08 (d, J = 2.3 Hz, 1H, ArH), 3.87 (t, J = 6.5 Hz, 2H, CH2), 3.30 (q, J = 7.1 Hz, 4H, CH2), 1.72–1.54 (m, 2H, CH2), 1.37–1.29 (m, 2H, CH2), 1.26–1.20 (m, 4H, CH2), 1.12 (t, J = 7.1 Hz, 6H, CH3), 0.80 (t, J = 6.8 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 181.71, 157.27, 150.76, 148.69, 139.96, 135.30, 129.61, 126.94, 118.03, 111.11, 102.65, 94.32, 67.16, 43.45, 30.44, 28.27, 24.72, 21.62, 13.03, 11.70 (MALDI) (M+, C23H31NO2S): calcd: 385.56; found: 385.74.:hexane = 1:10) to give compound 5c as an orange-red solid in 73.3% yield (0.56 g, 1.47 mmol). 1H NMR (400 MHz, CDCl3) δ 9.87 (s, 0.11H, CHO), 9.52 (s, 0.89H, CHO), 8.17 (s, 0.11H, CH), 7.79 (s, 0.89H, CH), 7.31 (d, J = 6.2 Hz, 1H, CH), 7.00 (dd, J = 5.1, 3.5 Hz, 1H, CH),6.98 (m, 1H, CH) 6.95 (dd, J = 8.6, 5.1 Hz, 1H, PrH), 5.98 (d, J = 2.0 Hz, 1H, PrH), 5.95 (m, 1H, PrH), 3.95 (t, J = 6.4 Hz, 2H, CH2), 3.27 (q, J = 7.1 Hz, 4H, CH2), 1.83–1.73 (m, 2H, CH2), 1.50–1.40 (m, 4H, CH2), 1.35–1.25 (m, 2H, CH2), 1.14 (t, J = 7.1 Hz, 0.66H, CH3), 1.09 (t, J = 7.1 Hz, 5.34H, CH3), 0.84 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 191.95, 159.53, 150.49, 146.18, 134.43, 130.66, 126.45, 126.20, 125.36, 109.68, 103.06, 93.01, 67.40, 43.38, 30.47, 28.16, 24.73, 21.46, 13.00, 11.66 (MALDI) (M+, C23H31NO2S): calcd: 385.56; found: 385.80.:hexane = 1:10) to give compound 4c as orange oil in 86.7% yield (0.47 g, 1.56 mmol). 1H NMR (400 MHz, CDCl3) δ 9.55 (d, J = 8.1 Hz, 1H, CHO), 7.70 (d, J = 15.4 Hz, 1H, CH), 7.39 (d, J = 8.4 Hz, 1H, PhH), 6.60 (dd, J = 15.4, 8.1 Hz, 1H, CH), 6.28 (d, J = 8.4 Hz, 1H, PhH), 6.09 (s, 1H, PhH), 4.01 (t, J = 5.2 Hz, 2H, CH2), 3.43 (q, J = 7.1 Hz, 4H, CH2), 1.92–1.77 (m, 2H, CH2), 1.49 (m, 4H, CH2), 1.40–1.32 (m, 2H, CH2), 1.21 (t, J = 7.1 Hz, 6H, CH3), 0.93–0.89 (t, J = 7.1 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 193.31, 159.18, 150.81, 148.30, 129.80, 122.68, 103.68, 93.72, 67.39, 43.71, 30.56, 28.19, 24.84, 21.55, 12.94, 11.69. MS (EI) (M+, C19H29NO2): calcd: 303.44; found: 303.23.:hexane = 1:5) to give chromophore A as a green solid in 46.7% yield (0.14 g, 0.23 mmol). 1H NMR (400 MHz, acetone) δ 8.26 (d, J = 9.2 Hz, 1H, PrH), 8.15 (d, J = 16.0 Hz, 1H, CH), 8.06 (s, 1H, CH), 7.74 (d, J = 4.1 Hz, 1H, CH), 7.30 (d, J = 4.1 Hz, 1H, CH), 6.88 (d, J = 16.0 Hz, 1H, CH), 6.53 (dd, J = 9.2, 2.3 Hz, 1H, PrH), 6.33 (d, J = 2.3 Hz, 1H, PrH), 4.17 (t, J = 6.2 Hz, 2H, CH2), 3.57 (q, J = 7.1 Hz, 4H, CH2), 1.90 (s, 6H, CH3), 1.64–1.52 (m, 2H, CH2), 1.47–1.36 (m, 4H, CH2), 1.34–1.27 (m, 2H, CH3), 0.92 (t, J = 7.1 Hz, 3H, CH3). 13C NMR (101 MHz, acetone) δ 178.18, 175.67, 161.74, 153.72, 151.07, 140.05, 139.19, 138.16, 137.63, 130.15, 126.30, 118.91, 114.04, 112.67, 111.68, 110.89, 106.30, 98.75, 95.21, 69.26, 45.28, 32.25, 26.94, 26.21, 24.10, 23.34, 14.37, 13.01 (MALDI) (M+, C35H37N5O2S): calcd: 591.77; found: 591.23. Anal. calcd (%) for C35H37N5O2S: C, 71.04; H, 6.30; N, 11.83; found: C, 71.09; H, 6.37; N, 11.79.Acknowledgements
We are grateful to the National Natural Science Foundation of China (no. 11104284 and no. 61101054) for the financial support.Notes and references
- J. Y. Lee, H. B. Bang, T. S. Kang and E. J. Park, Eur. Polym. J., 2004, 40, 1815–1822 CrossRef CAS PubMed.
- B. J. Coe, S. P. Foxon, E. C. Harper, M. Helliwell, J. Raftery, C. A. Swanson, B. S. Brunschwig, K. Clays, E. Franz, J. Garin, J. Orduna, P. N. Horton and M. B. Hursthouse, J. Am. Chem. Soc., 2010, 132, 1706–1723 CrossRef CAS PubMed.
- I. D. L. Albert, T. J. Marks and M. A. Ratner, J. Am. Chem. Soc., 1997, 119, 6575–6582 CrossRef CAS.
- E. Koscien, J. Sanetra, E. Gondek, A. Danel, A. Wisla and A. V. Kityk, Opt. Commun., 2003, 227, 115–123 CrossRef CAS PubMed.
- Q. Q. Li, C. G. Lu, J. Zhu, E. Fu, C. Zhong, S. Y. Li, Y. P. Cui, J. G. Qin and Z. Li, J. Phys. Chem. B, 2008, 112, 4545–4551 CrossRef CAS PubMed.
- W. Gong, Q. Q. Li, Z. Li, C. G. Lu, J. Zhu, S. Y. Li, J. W. Yang, Y. P. Cui and J. G. Qin, J. Phys. Chem. B, 2006, 110, 10241–10247 CrossRef CAS PubMed.
- H. Kang, G. Evmenenko, P. Dutta, K. Clays, K. Song and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 6194–6205 CrossRef CAS PubMed.
- Y. Liao, S. Bhattacharjee, K. A. Firestone, B. E. Eichinger, R. Paranji, C. A. Anderson, B. H. Robinson, P. J. Reid and L. R. Dalton, J. Am. Chem. Soc., 2006, 128, 6847–6853 CrossRef CAS PubMed.
- J. A. Davies, A. Elangovan, P. A. Sullivan, B. C. Olbricht, D. H. Bale, T. R. Ewy, C. M. Isborn, B. E. Eichinger, B. H. Robinson, P. J. Reid, X. Li and L. R. Dalton, J. Am. Chem. Soc., 2008, 130, 10565–10575 CrossRef CAS PubMed.
- B. H. Robinson and L. R. Dalton, J. Phys. Chem. A, 2000, 104, 4785–4795 CrossRef CAS.
- F. Dumur, C. R. Mayer, E. Dumas, F. Miomandre, M. Frigoli and F. Secheresse, Org. Lett., 2008, 10, 321–324 CrossRef CAS PubMed.
- R. M. El-Shishtawy, F. Borbone, Z. M. Al-Amshany, A. Tuzi, A. Barsella, A. M. Asiri and A. Roviello, Dyes Pigm., 2013, 96, 45–51 CrossRef CAS PubMed.
- A. Leclercq, E. Zojer, S. H. Jang, S. Barlow, V. Geskin, A. K. Y. Jen, S. R. Marder and J. L. Bredas, J. Chem. Phys., 2006, 124, 7 CrossRef PubMed.
- X. H. Ma, F. Ma, Z. H. Zhao, N. H. Song and J. P. Zhang, J. Mater. Chem., 2010, 20, 2369–2380 RSC.
- P. R. Varanasi, A. K. Y. Jen, J. Chandrasekhar, I. N. N. Namboothiri and A. Rathna, J. Am. Chem. Soc., 1996, 118, 12443–12448 CrossRef.
- Z. S. Wang, Y. Cui, Y. Dan-Oh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. C, 2008, 112, 17011–17017 CAS.
- Z. S. Wang, Y. Cui, K. Hara, Y. Dan-Oh, C. Kasada and A. Shinpo, Adv. Mater., 2007, 19, 1138–1141 CrossRef CAS.
- Y. Z. Wu and W. H. Zhu, Chem. Soc. Rev., 2013, 42, 2039–2058 RSC.
- M. J. Cho, S. K. Lee, J. I. Jin and D. H. Choi, Macromol. Res., 2006, 14, 603–609 CrossRef CAS.
- M. J. Cho, J. H. Lim, C. S. Hong, J. H. Kim, H. S. Lee and D. H. Choi, Dyes Pigm., 2008, 79, 193–199 CrossRef CAS PubMed.
- K. Staub, G. A. Levina, S. Barlow, T. C. Kowalczyk, H. S. Lackritz, M. Barzoukas, A. Fort and S. R. Marder, J. Mater. Chem., 2003, 13, 825–833 RSC.
- H. J. Xu, M. L. Zhang, A. R. Zhang, G. W. Deng, P. Si, H. Y. Huang, C. C. Peng, M. K. Fu, J. L. Liu, L. Qiu, Z. Zhen, S. H. Bo and X. H. Liu, Dyes Pigm., 2014, 102, 142–149 CrossRef CAS PubMed.
- C. Zhang, L. R. Dalton, M. C. Oh, H. Zhang and W. H. Steier, Chem. Mater., 2001, 13, 3043–3050 CrossRef CAS.
- X. Q. Piao, X. M. Zhang, S. Inoue, S. Yokoyama, I. Aoki, H. Miki, A. Otomo and H. Tazawa, Org. Electron., 2011, 12, 1093–1097 CrossRef CAS PubMed.
- J. Y. Wu, J. L. Liu, T. T. Zhou, S. H. Bo, L. Qiu, Z. Zhen and X. H. Liu, RSC Adv., 2012, 2, 1416–1423 RSC.
- C. Lee and R. G. Parr, Phys. Rev. A, 1990, 42, 193–200 CrossRef CAS.
- C. M. Isborn, A. Leclercq, F. D. Vila, L. R. Dalton, J. L. Bredas, B. E. Eichinger and B. H. Robinson, J. Phys. Chem. A, 2007, 111, 1319–1327 CrossRef CAS PubMed.
- R. M. Ma, P. Guo, L. L. Yang, L. S. Guo, X. X. Zhang, M. K. Nazeeruddin and M. Gratzel, J. Phys. Chem. A, 2010, 114, 1973–1979 CrossRef CAS PubMed.
- S. T. Huang, Y. C. Hsu, Y. S. Yen, H. H. Chou, J. T. Lin, C. W. Chang, C. P. Hsu, C. Tsai and D. J. Yin, J. Phys. Chem. C, 2008, 112, 19739–19747 CAS.
- R. V. Solomon, P. Veerapandian, S. A. Vedha and P. Venuvanalingam, J. Phys. Chem. A, 2012, 116, 4667–4677 CrossRef CAS PubMed.
- J. Y. Wu, S. H. Bo, J. L. Liu, T. T. Zhou, H. Y. Xiao, L. Qiu, Z. Zhen and X. H. Liu, Chem. Commun., 2012, 48, 9637–9639 RSC.
- K. S. Thanthiriwatte and K. M. N. de Silva, J. Mol. Struct.: THEOCHEM, 2002, 617, 169–175 CrossRef CAS.
- X. H. Zhou, J. Davies, S. Huang, J. D. Luo, Z. W. Shi, B. Polishak, Y. J. Cheng, T. D. Kim, L. Johnson and A. Jen, J. Mater. Chem., 2011, 21, 4437–4444 RSC.
- L. T. Cheng, W. Tam, S. H. Stevenson, G. R. Meredith, G. Rikken and S. R. Marder, J. Phys. Chem., 1991, 95, 10631–10643 CrossRef CAS.
- L. T. Cheng, W. Tam, S. R. Marder, A. E. Stiegman, G. Rikken and C. W. Spangler, J. Phys. Chem., 1991, 95, 10643–10652 CrossRef CAS.
- J. K. Gao, Y. J. Cui, J. C. Yu, Z. Y. Wang, M. Q. Wang, J. R. Qiu and G. D. Qian, Macromolecules, 2009, 42, 2198–2203 CrossRef CAS.
- C. C. Teng and H. T. Man, Appl. Phys. Lett., 1990, 56, 1734–1736 CrossRef CAS PubMed.
- D. H. Park, C. H. Lee and W. N. Herman, Opt. Express, 2006, 14, 8866–8884 CrossRef.
- I. V. Kityk, M. Makowska-Janusik, E. Gondek, L. Krzeminska, A. Danel, K. J. Plucinski, S. Benet and B. Sahraoui, J. Phys.: Condens. Matter, 2004, 16, 231–239 CrossRef CAS.
- L. R. Dalton, P. A. Sullivan, D. H. Bale and B. C. Bricht, Solid-State Electron., 2007, 51, 1263–1277 CrossRef CAS PubMed.
- J. Wu, C. Peng, H. Xiao, S. Bo, L. Qiu, Z. Zhen and X. Liu, Dyes Pigm., 2014, 15–23 CrossRef PubMed.
- M. A. Mortazavi, A. Knoesen, S. T. Kowel, B. G. Higgins and A. Dienes, J. Opt. Soc. Am. B, 1989, 6, 733–741 CrossRef CAS.
- Y. H. Yang, H. J. Xu, F. G. Liu, H. R. Wang, G. W. Deng, P. Si, H. Y. Huang, S. H. Bo, J. L. Liu, L. Qiu, Z. Zhen and X. H. Liu, J. Mater. Chem. C, 2014, 2, 5124–5132 RSC.
- M. Q. He, T. M. Leslie and J. A. Sinicropi, Chem. Mater., 2002, 14, 2393–2400 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |