
Microfluidics & nanotechnology: towards fully integrated analytical devices for the detection of cancer biomarkers
G. Perozzielloab,
P. Candeloroa,
F. Gentilea,
A. Nicastric,
A. Perric,
M. L. Coluccioa,
A. Adamod,
F. Pardeoa,
R. Catalanoa,
E. Parrottac,
H. D. Espinosab,
G. Cuda*c and
E. Di Fabrizio*ae
aBioNEM lab, Department of Experimental and Clinical Medicine, University “Magna Graecia” of Catanzaro, Catanzaro, 88100, Italy. E-mail: enzo.difabrizio@kaust.edu.sa; Fax: +9667035278358; Tel: +966128084307
bDepartment of Mechanical Engineering, NorthWestern University, Evanston, 60208, IL, USA
cProteomics lab, Department of Experimental and Clinical Medicine, University “Magna Graecia” of Catanzaro, Catanzaro, 88100, Italy. E-mail: cuda@unicz.it; Fax: +3909613694073; Tel: +3909613694225
dDepartment of Chemical Engineering, Massachusetts Institute of Technology (MIT), Cambridge, MA 02139-4307, USA
eKing Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Kingdom of Saudi Arabia
First published on 21st October 2014
Abstract
In this paper, we describe an innovative modular microfluidic platform allowing filtering, concentration and analysis of peptides from a complex mixture. The platform is composed of a microfluidic filtering device and a superhydrophobic surface integrating surface enhanced Raman scattering (SERS) sensors. The microfluidic device was used to filter specific peptides (MW 1553.73 D) derived from the BRCA1 protein, a tumor-suppressor molecule which plays a pivotal role in the development of breast cancers, from albumin (66.5 KD), the most represented protein in human plasma. The filtering process consisted of driving the complex mixture through a porous membrane having a cut-off of 12–14 kD by hydrodynamic flow. The filtered samples coming out of the microfluidic device were subsequently deposited on a superhydrophobic surface formed by micro pillars on top of which nanograins were fabricated. The nanograins coupled to a Raman spectroscopy instrument acted as a SERS sensor and allowed analysis of the filtered sample on top of the surface once it evaporated. By using the presented platform, we demonstrate being able to sort small peptides from bigger proteins and to detect them by using a label-free technique at a resolution down to 0.1 ng μL−1. The combination of microfluidics and nanotechnology to develop the presented microfluidic platform may give rise to a new generation of biosensors capable of detecting low concentration samples from complex mixtures without the need for any sample pretreatment or labelling. The developed devices could have future applications in the field of early diagnosis of severe illnesses, e.g. early cancer detection.
Introduction
The growing interest in personalized medicine requires rapid, sensitive and low cost methodologies for the analysis of compounds, which can be correlated to diseases. In the last years, proteomics is becoming a multidisciplinary field, which shows the potential to have the aforementioned requirements. In fact, proteins and peptides are used as biomarkers for the most crucial and important diseases.1–3 The investigation of biomarkers currently involves the analysis of biological samples by chromatographic and mass spectrometry techniques. These include slow, expensive and invasive procedures, and are not sensitive enough to complete the analysis of complex samples where the target molecules are present in low concentrations. Nowadays there are analytical techniques that, starting from a sample with a few molecular components, allow the detection and characterization of the individual components at the level of single molecules. These techniques, however, fail when the “mixture” becomes complex, such as the blood plasma. In these cases, it is quite difficult to discriminate the various components. Conventional tools and technologies to purify the sample are based on solid-phase extraction (SPE), “ultrafiltration”, “acetonitrile precipitation”, “size-exclusion chromatography” and “SDS-PAGE”, “immuno affinity chromatography” or others. These methods require complicated and expensive equipment, which needs manual labor, and are often time-consuming. Therefore, there is a great need to increase the efficiency and speed of these procedures by reducing the number of manual operations and the use of expensive reagents. Microfluidic devices can integrate different microstructures4–7 for the extraction and purification of the sample and have the advantage of speeding up these types of operations, significantly lowering the processing costs.8–11 Microfluidic extraction and purification methods have been explored by several research groups. Depending on the type of material to be isolated, a variety of techniques can be considered, such as the use of mechanical filters that separate according to size of the substances,12 filters based on the exploitation of the principles of fluid dynamics,13 dielectrophoresis,14 electrophoresis,15 magnetophoresis or magnetic traps16 or by biofunctionalised substrates.17 Finally, in many cases, the molecules of interest present in a complex biological sample, are present in such low concentrations that cannot be analyzed using current techniques, such as mass spectrometry (ESI, SLD, MALDI), protein microarray, ELISA, Western blotting, interferometry. Several groups have developed methods capable of analyzing single molecules based on surface acoustic wave propagation,18 nanopore-based sensors,19 fluorescence microscopy,20 extended or localized surface plasmon resonance (SPR),21–24 surface enhanced Raman scattering (SERS)25,26 and diffraction.27 Among the optical biosensors, those exploiting surface plasmons are the most common. The renewed interest in plasmon comes mainly from the ability to concentrate and to propagate the light in structures smaller than the wavelength of the probe, thus ensuring well-focused spots below the diffraction limit.28–30 In addition, this way to concentrate the light, in many cases, has an interesting side effect, that is an increase of high electromagnetic field. This improvement can be used to influence the interactions of light with matter and to increase the non-linear phenomena at a detectable level.31–33 In recent years, it has also aroused much interest in using nanotechnology and nanodevices coupled with microfluidics especially in in vitro biological tests and practices.34,35 This brings more and more towards a fully integration of analytical devices, where all the previously mentioned operations are performed on a single platform. In this work, we propose the implementation of a modular microfluidic platform able to pretreat complex mixtures of biological samples, isolate peptides of interest and to integrate plasmonic nanodevices for analysis of very low concentrated biological compounds. The platform is composed of three modules: – a microfluidic device integrating a filter membrane which can separate proteins and peptides by size; – a superhydrophobic surface which is used to concentrate the separated sample on top of plasmonic sensors integrated on the same surface, which consisted of silver nanograins nanostructured on top of the pillars, being able to reproduce SERS effects; – a Raman spectroscopy instrument coupled to the SERS surfaces which allows to detect the biological sample. Experiments were performed to detect a synthetic peptide, VTREWVLDSVALYQCQ, which was designed based on a specific portion of the BRCT (BRCA1 carboxyl-terminal) sequence domain of the BRCA1 protein (Breast Cancer Associated gene 1), which plays a pivotal role in the development of the breast cancer. In the following section: (i) we describe how the platform was fabricated and characterized; (ii) provide details of experimental protocols set to perform analysis of peptides present at very low concentrations, and, (iii) validate the procedure by showing the ability of this platform to separate peptides from albumin and identify the separated sample even if present at very low concentration (down to 0.1 ng μL−1).Materials and methods
Materials
Superhydrophobic substrate fabrication
Artificial super-hydrophobic surfaces (SHSs) were fabricated following the methods reported in.40 SHSs are textures comprising a periodic hexagonal lattice of cylindrical Si micro-pillars with a specific diameter and pitch. (100) silicon wafers were cleaned with acetone and isopropanol to remove possible contaminant and then etched with a 4% wet HF solution. The wafers were then rinsed with DI water and dried with N2. Standard optical lithography techniques (Karl Suss Mask Aligner MA 45, SussMicroTec GA, Garching, Germany) were used to obtain regular arrays of disks within a layer of positive resist (S1813, from Rohm and Haas) that was spin-coated onto clean silicon wafers. Therefore, electroless deposition techniques were employed to grow silver nano-grains within the holes. Electroless plating is a site selective deposition technique in which ions are deposited as metals on specific sites of a silicon substrate to obtain clusters of metallic nanoparticles with the desired characteristics.40 A fluoridric acid (HF) solution containing silver nitrate (AgNO3) was used, where Ag was reduced to metal form by the Si substrate oxidation. In particular, the patterned silicon wafer was dipped in a 0.15 M HF solution containing 1 mM silver nitrate for 60 s at a constant temperature T = 313 K. After the growth process, the silicon wafer was rinsed with water and dried under nitrogen flux. Upon removal of the residual resist with acetone, a Bosch Reactive Ion Etching (MESC Multiplex ICP, STS, Imperial Park, Newport, UK) process was utilized whereby the final structures were obtained in the form of cylindrical pillars with a diameter d = 10 μm, a pitch δ = 30 μm, and an aspect ratio greater than 2. The substrates, as a whole, were then covered with a thin (few nm) film of a Teflon-like (C4F8) polymer to ensure hydrophobicity; to do this, a modified Bosch/RIE process was utilized, where solely the passivation mode was activated. In this phase, all gas flows, including SF6, argon, and oxygen, are set to zero, with the exception of the chemical inert passivation layer C4F8. The masks necessary for optical lithography were fabricated using standard electron beam lithography (Crestec CABL-9000C electron beam lithography system) methods.Superhydrophobic substrate characterization
SEM images of the SHSs were captured using a Dual Beam (SEM-FIB)-FEI Nova 600 Nano Lab system. During the acquisitions, beam energies of 5 and 15 keV, and corresponding electron currents of 0.98 pA and 0.14 nA, were used. In some cases, the mode 2 configuration was used, whereby images can be magnified over 2500k× and ultrahigh resolution may be achieved. In this modality, the immersion lens was switched on. Moreover, the superhydrophobic surfaces were tested by performing contact angle measurements to characterize the hydrophobicity level. An automatic contact angle meter (KSV CAM 101, KSV Instruments) was used at room temperature.Please notice that the energy of adhesion γ per unit area at the gas/water interface is ∼72.8 mJ m−2 at 20 °C.
Microfluidic device fabrication
The devices were made of two layers of poly-methyl-methacrylate (PMMA) (3 cm long, 2 cm wide and 0.8 mm high) machined by micromilling (MiniTech®) in order to reproduce on the different layers the microfluidic chambers and channels, and a membrane. The two layers of PMMA and the membrane were then bonded together (see Fig. 1). In particular the top layer integrates the upper micro chamber, inlet and outlet channels; the bottom layer integrates the bottom micro chamber and inlet and outlet channels. The microchamber has a square shape 3 mm in side and 0.1 mm deep. The microfluidic channels have a cross section of 0.2 mm in depth and 0.25 mm in width. The bonding process was an UV light (Dymax®, 400 W at 365 nm) assisted bonding. This process consists in placing the PMMA layers under UV (for 90 seconds). The PMMA layers and the membrane are then assembled together, pressed (at 8 kN) at a temperature of 85 °C for 1 hour.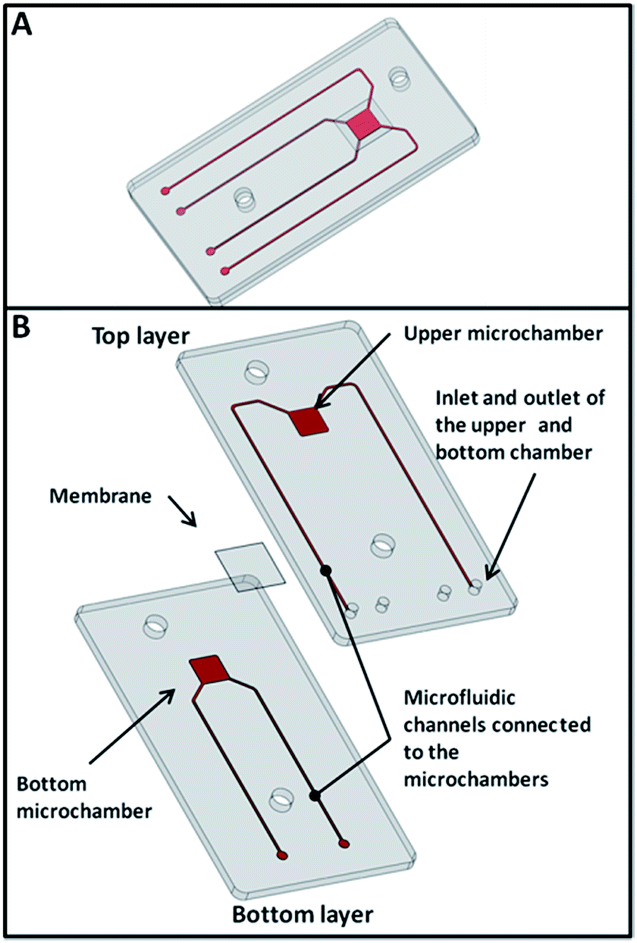 | ||
| Fig. 1 CAD drawing of the microfluidic device: isometric view (A) and exploded view (B). | ||
Microfluidic device characterization
The device was characterized implementing and testing different microfluidic protocols. For this purpose, it was positioned on a stage of a microscope (Nikon Eclipse TE2000-U) equipped with a CCD camera (Nikon DS-2Mw) connected to a PC in order to monitor liquid flows and absence of leakages. Bonding tests were also performed for detecting the maximum pressure at which the microfluidic device could operate without leakages or breakages of the devices.Raman measurement set-up
The nanograins integrated in correspondence of the superhydrophobic surface pillars allowed to perform SERS measurements on the peptides which were filtered into the microfluidic device, deposited on the superhydrophobic surface and there evaporated. The nanograins were coupled to a Raman micro-spectroscopy equipment “Alpha 300RA” from Witec Company. The laser source was a 532 nm laser coupled through a monomodal optical fiber to the microscope; the incident light was focused on the sample by means of a 100×/0.95 NA objective and the laser power at the sample level was kept very low, in the range of 0.1–0.5 mW. Indeed, due to SERS effect of the Ag nanograins, larger laser power could result in a photo damaging of the probed sample. The choice of a 532 nm laser was dictated by the plasmon resonances of Ag nanoparticles, occurring at these wavelengths. The backscattered light was collected through the same objective, filtered out of the Rayleigh peak by a notch filter, and sent to an 1800 lines per mm grating for spectral analysis. The probed spectral interval was from 1200 cm−1 up to 1800 cm−1, while the integration time range was between 0.5 and 2.0 s.Experimental protocols
The experimental protocol consisted in purifying a biological sample in the microfluidic device. The filtrated sample was then transported from the output of the microfluidic device on top of the superhydrophobic surface, where it was left to evaporate. The dry sample was then analyzed by a Raman spectroscopy instrument. Fig. 2 (top) shows a scheme of the experimental protocol. The experiments were conducted at least three times on: (1) a sample containing the peptide at a concentration of 10 ng μL−1 and albumin (35 μg μL−1) diluted in DI water; (2) samples containing the peptide diluted in DI water at the following concentrations (1 μg μL−1, 1 ng μL−1, and 0.1 ng μL−1). The samples described in (2) were used as control and to prove the high resolution of analysis, which can be reached, by using this platform. The experiments, in which the sample described in (1) was used, were repeated after washing the microfluidic device with a specific protocol described in the following section. Raman measurements were also taken on a pure peptide (1 μg μL−1) diluted in DI water which was placed on top of the superhydrophobic surface and was left to evaporate. This measurement was used as control.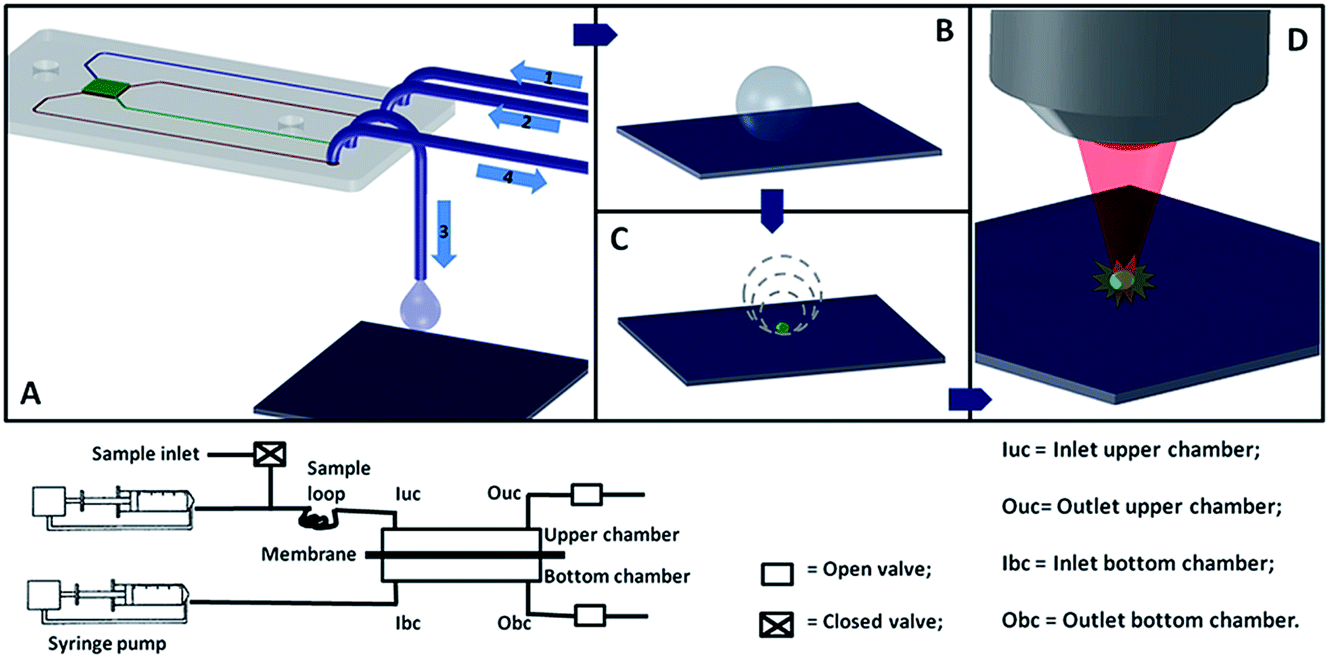 | ||
| Fig. 2 (top) schematic drawing showing the experimental protocols to: (A) filter the biological sample in the microfluidic device, the sample is injected through the channel 1, it is forced to go through the membrane which is placed in the microchamber (at the center of the microfluidic device) and it is recovered from the channel 3, the device membrane can then be cleaned for a successive use by washing DI water from the channel 2 to the channel 4 which is connected with the waste; (B and C) concentrate the filtrated sample on top of the superhydrophobic surface; (D) analyse the concentrated sample by coupling a Raman spectroscopy instrument with the superhydrophobic surface and performing SERS measurements; (bottom) scheme of the microfluidic circuit connected to the microfluidic device. | ||
Results
Microfluidic device for sample filtering
Before using it, the microfluidic device was tested to investigate the bonding strength. To this end, the microfluidic device was connected to a pressure source and the outlets were obstructed. The device was then immersed in water for observing bubble formation when the pressure was increased. No bubbles were observed by using pressures up to 1.5 bar. In Fig. 3a, it is possible to observe a picture of the microfluidic device connected to syringe pumps through external tubes and an external frame. The inlet of the upper chamber of the microfluidic device is connected to a sample port and a sample loop. A valve allows switching on and off the sample port. The outlet of the microfluidic device are connected to valves which allow to direct the flows in the upper or the bottom chamber allowing to perform operation of filtering or cleaning of the device. Fig. 3B shows a close-up of the microfluidic device. The specific layout of the microfluidic chambers and channels was designed to allow the assembling of the same device by using two layers only of PMMA and the filtering membrane. In fact, the microfluidic channels never meet each other during their path, thus there was no need of any interlayer between the channels connected to the upper chamber and those connected to the bottom chamber. Fig. 3C represents a SEM image of the filtering membrane demounted from a microfluidic system that was used to filter albumin. It can be clearly seen the albumin sticking on top of the membrane, unable to pass through it. | ||
| Fig. 3 (A) Photograph of the microfluidic device connected to the syringe pumps through an external frame and coupled to an optical microscope; (B) close-up of the microfluidic device in which: 1 is the inlet for the upper microfluidic chamber, 2 is the inlet of the bottom microfluidic chamber, 3 is the outlet of the bottom microfluidic chamber, 4 is the outlet of the upper microfluidic chamber, 5 indicates the upper and the bottom microfluidic chamber divided by the filtering membrane; (C) represents a SEM image of the filtering membrane demounted from the microfluidic chamber once it was used. | ||
Superhydrophobicity of the micro and nanostructured substrate
In Fig. 4A, it can be observed micropillars etched on silicon substrates following the fabrication process described in the previous section to realize superhydrophobicity effects on a surface. | ||
| Fig. 4 SEM images of: micropillars etched on silicon favoring super-hydrophobicity effects (A); zoom-in of a pillar showing the silver nanograins, which allow to reproduce SERS effects during Raman analysis (B and C); picture taken at the contact angle meter of a liquid drop placed on top of the super-hydrophobic surface (D). Zoom-in of an area in which a biological sample was concentrated on top of the pillars (E and F). | ||
In particular, Fig. 4B and C shows a zoom-in of the silver nanograins deposited on top of the micropillars by means of the electroless process. The electroless grown Ag layer has about 1 particle diameter, where the particle diameter is roughly 50 nm with a standard deviation S.D. = 10 nm. The particles in the cluster distribute following a Gaussian curve. This layer served as mask during the reactive ion etching (RIE) process, while its characteristic granular structure allowed for the enhancement of the SERS signal. In fact, nano-sized geometries appropriately positioned upon the pillars would ensure giant SERS enhancement during Raman spectroscopy analysis. As reported earlier,44 Ag nanoparticles experience localized plasmon resonances when illuminated with laser light of suitable wavelength. Plasmon resonances and plasmon oscillations are collective vibrations of the electron gas within metal nanoparticles. As concerned with oscillating electrical charges, these phenomena are accompanied by oscillating electromagnetic fields able to interact with external, far-field radiations. In particular, the far-field radiation excites localized plasmon resonance, which in turn enhances the local electromagnetic field close to the nanoparticle surface.
This enhanced field causes an improved Raman signal from the molecules attached to the Ag surface, and this latter signal is detected as an enhanced Raman scattering in the far field region. The result is the well-known surface enhanced Raman scattering (SERS), which largely improves Raman spectroscopy technique. It has to be mentioned that the C4F8 layer is few atomic layers thick only, that is, in the very low nanometer range. Therefore, it does not alter the Raman effect, and this has been demonstrated in.40 Fig. 5D shows a drop of the biological sample deposited on top of the superhydrophobic surface. It is clear the hydrophobic effect that conveys the liquid drop to have a contact angle of 170° with the surface. The SEM image shown in Fig. 4E and F demonstrate how the filtrated peptides, present in the initial biological sample at low concentration (see sections above) were concentrated on top of few pillars after the evaporation of the drop and are ready for being analyzed by Raman spectroscopy.
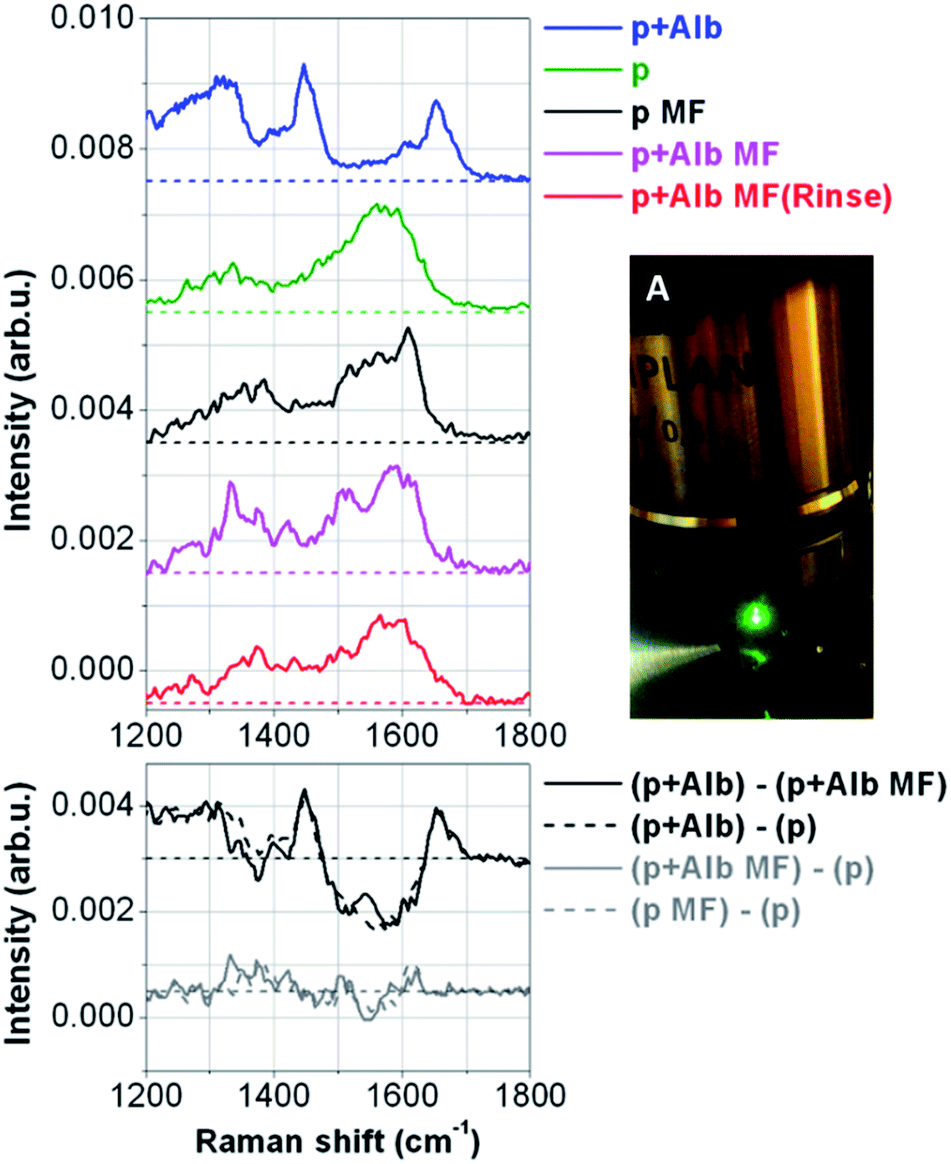 | ||
| Fig. 5 (A) Raman spectroscopy objective focusing on a biological sample on top of a superhydrophobic surface; left top: Raman spectra measurements on the peptide (1 μg μL−1) mixed with albumin (35 μg μL−1) (p + alb) (not filtered sample), pure peptide (1 μg μL−1) (p), pure peptide filtered in the microfluidic device (p MF), peptide (1 ng μL−1) mixed with albumin (35 μg μL−1) filtered in the microfluidic device (p + Alb MF), peptide (1 ng μL−1) mixed with albumin (35 μg μL−1) filtered in the microfluidic device after having performed a washing protocol (p + ALB MF (Rinse)); left bottom: spectra difference of the peptide mixed with Albumin minus the peptide mixed with albumin filtered in the microfluidic device ((p + Alb) − (p + Alb MF)), the peptide mixed with albumin minus the pure peptide ((p + Alb) − (p)), the peptide mixed with albumin filtered in the microfluidic device minus the pure peptide ((p + Alb MF) − (p)), the pure peptide filtered in the microfluidic device minus the pure peptide ((p MF) − (p)). | ||
Sample concentration on the superhydrophobic substrates
Small drops of deionized water, containing the moieties at study, were positioned upon the substrate and let it to evaporate. In sight of a simple balance of forces, the line of contact at the solid interface would therefore recede with time and thus the footprint of the drop would gradually reduce. When the drop gets sufficiently small (where “small” is defined on the basis of a mathematical criterion described in ref. 44 and depends on the geometry and surface chemistry of the substrate), a transition to a more stable state occurs, whereby the drop is firmly attached to the substrate, and the scale-down of the area of contact is prevented. Thereupon, few molecules could be accumulated in a very small region, assuring an increased density and, accordingly, the attainment of the limits of detection.Raman measurements on filtrated and concentrated samples
Fig. 5(A) shows the Raman Spectroscopy objective, which focus the green laser on top of the superhydrophobic surface in correspondence of a spot where the filtered peptides were evaporated. Here it can be noticed drops of samples that were still not evaporated having a perfect spherical shape (three small drops on the right of the laser spot). Raman spectra were recorded on control samples, which were the peptide and albumin, dissolved together in DI water at the concentration of respectively 1 μg μL−1 and 35 μg μL−1 (not filtered sample) and the pure peptide dissolved in water (1 μg μL−1) evaporated on the superhydrophobic surface. Moreover, measurements were taken from samples which were filtered in the microfluidic device, concentrated and evaporated on the superhydrophobic surface which were the pure peptide (1 ng μL−1) used as control, the peptide mixed with albumin at the same concentration showed above, and the same sample filtered after having performed the washing protocol of the device. The results are shown in Fig. 5 (left). The first curve from the top is the Raman spectrum recorded on the sample of mixed peptide and albumin (p + Alb). It is mainly dominated by the large Raman signal coming from albumin, due to the large concentration of albumin in the solution but also due to the much larger number of aminoacid residues of albumin compared to the short sequence of the peptide. The most evident peaks are then the 1445 cm−1 band due to CH bond deformation,41 and the 1650 cm−1 that is the so-called Amide I vibration due to the CO stretching.42 The bottom curves are all recorded on peptide samples after different treatments: from the second curve to the bottom one, we have the spectrum of the pure peptide (p), the pure peptide after microfiltration (p MF) for showing that pure peptide is not affected by the microfluidic treatment (p + Alb MF), peptide mixed with albumin after microfiltration (magenta), and finally peptide mixed with albumin after microfiltration performed after a rinsing of the microfluidic device (p + Alb MF rinse). While, at first glance, the spectra where the peptides were measured look slightly dissimilar each other, it can be clearly seen some similarities by looking at the broad peaks at about 1350 cm−1 and 1560 cm−1. These two bands are likely due to in plane C–C vibrations (the latter one),43 while the former one is a typical frequency of tryptophan residues,44 which have larger Raman scattering cross-sections compared to the other amino acids of the peptide sequence. The slight differences of these bands were due to different local SERS effects caused by the random configuration of the silver nanograins on top of the superhydrophobic surface. The result, which is more evident, is that the spectra obtained by the measurements on filtered samples have all the same trend of the spectrum obtained from the measurements on the pure peptide. It is also important to notice that the characteristic peaks of the albumin present on the measurements taken on the not filtered sample, at 1445 and 1650 cm−1, disappear in the spectra obtained from the filtered samples. This is a clear proof that the albumin was filtered and that it was possible to measure single peptides from a complex mixture. Once the microfluidic device was used, it was cleaned by using a washing procedure and then used again to filter a new sample, which was subsequently placed on the superhydrophobic substrate, evaporated and measured. The spectrum measured from such a sample is shown in Fig. 5, it can be clearly seen that it is similar to that obtained by the measurements taken on the first processed sample. This result demonstrates that the microfluidic platform can be used continuously for more experiments without the need to exchange the microfluidic filter. In the bottom panel of the same figure, we show some significant difference spectra. First, we have the difference spectrum between the sample of the mixed peptide with albumin (p + Alb) and the same sample after microfiltration (p + Alb MF). As we can see, the corresponding difference curve shows the characteristic peaks of albumin (1445 and 1650 cm−1) and a broad dip corresponding to the main band of the peptide (1560 cm−1). For comparison we also show the difference spectrum of peptide mixed with albumin (p + Alb) from the pure peptide (p), and the behaviour of the curve is the same as before, thus further proving that after microfiltration the peptide mixed with albumin has been cleaned from the albumin, and only the peptide is left.The last two curves are instead the difference spectra between peptide mixed with albumin after microfiltration (p + Alb MF) and the pure peptide (p), and the difference spectrum of pure peptide after microfiltration (p MF) and pure peptide (p). In both these cases we expect nearly flat curves (like the ones in the graph) without any pronounced peak or band, since after microfiltration we have only the peptide left, unaltered from the microfiltration itself, both for the peptide mixed with albumin (p + Alb) and for the simple peptide (p). Finally, we show the results achieved on peptide low concentrations using the combination of drop evaporation on superhydrophobic surface with SERS analysis (Fig. 6). On the top panel the spectra recorded on two samples with starting dilutions of 0.1 ng μL−1 of the peptide are shown. The two curves are similar each other, and they resemble the curves shown in Fig. 5. The signal to noise ratio is slightly worse due to the smaller concentration of the peptide, but they are still observable the two characteristic broad band at 1560 cm−1 and at 1350 cm−1, this latter one having the shape of a shoulder on the overall curve. Furthermore, we performed a xy scanning measurement over the top of the pillars, recording Raman spectra with a spatial step size of 2.0 μm. As a result, we show the pseudo-color map of the peak area in the range 1520–1610 cm−1 as a function of the spatial coordinates. We can clearly see that the most intense signals are recorded over the pillars, where the Ag nanograins responsible of the SERS effect are located. All around the signal has negligible band intensities, even if the optical picture of the scanned area (middle panel) shows that a continuous drop residual is suspended between the pillars. This evidence highlights the crucial role played by the Ag nanograins and SERS effect. Again, the differences observed between the signal intensities over the pillars is mainly due to a random size distribution of Ag nanograins, with the smallest nanograins acting as hot spots for the detected signal.
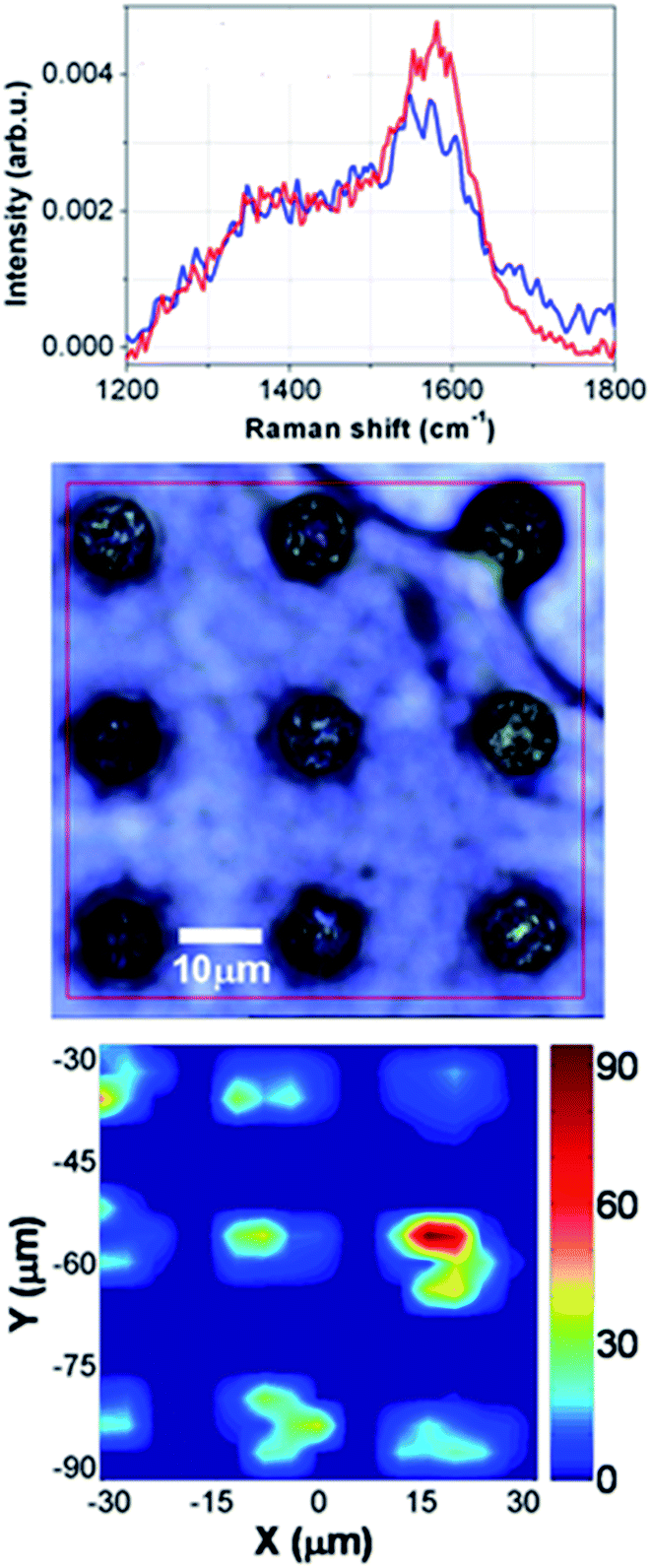 | ||
| Fig. 6 Top: Raman spectra measurements of two filtered samples where the peptides were present at a concentration 0.1 ng μL−1; middle: Optical view from the top of the micropillars where the proteins have been deposited; bottom: xy scanning Raman measurements over the top of the pillars. | ||
Conclusions
In conclusion, we developed a microfluidic platform consisting of three modules: a microfluidic device, a superhydrophobic surface integrating plasmonic structures and a Raman spectroscopy instrument coupled to the plasmonic structures. In particular the microfluidic device, fabricated in PMMA by micromilling and solvent assisted bonding, consisted of an upper chamber and a bottom chamber separated by a filtering membrane having a cut-off of 12–14 kD. The superhydrophobic surface was fabricated in silicon by optical lithography and plasma etching. It integrates micropillars on top of which nanograins are fabricated by an electroless deposition technique. The nanograins coupled to a Raman spectroscopy instrument acts as a SERS sensor allowing to perform label-free analysis of biological samples. The developed platform was used to analyse biological complex mixtures. In particular, it was used to filter a specific peptide, a BRCA1 fragment that is a breast cancer biomarker, from albumin and analyse it. The complex sample was injected in the microfluidic device and forced through the membrane to separate the peptides from the albumin. The filtered sample was then deposited on top of the superhydrophobic surface and, after evaporation, it was analysed by Raman spectroscopy exploiting the SERS effect of the nanograins. We demonstrated by using a simple protocol to effectively pretreat the sample in order to reach the sensing surface in ideal condition to be analysed. We detected peptide concentrations as low as 0.1 ng μL−1 from a complex mixture.Author contributions
GP designed the microfluidic device, the experimental plan, realized the microfluidic filtration experiments, supervised the experimental part of the work and wrote the manuscript. F.G. designed the superhydrophobic surface, the experimental plan, supervised the part of the work concerning the sample concentration, helped in realizing the experiments and wrote the manuscript. P.C. prepared the Raman set-up, realized the Raman measurements of the biological sample, interpreted the Raman data and wrote the manuscript. A.N., A.P., and E.P. helped in preparing the biological sample and in realizing the experiments. M.L.C. helped in preparing the solutions and supervised the part of the work concerning the microfiltration. F. P and R. C. took care of the microfluidic device fabrication and helped in performing the experiments of microfiltration. H.D.E. and A.A. supervised the part of the work concerning microfluidics and wrote the manuscript. G.C. supervised the part of the work concerning the biological samples, the sample treatment and analysis and E.D.F. designed the experimental plan, supervised the work and wrote the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was partially supported by the European project EUROMBR (grant no. 608104), Cariplo Foundation under the project “New Frontiers in Plasmonic Nanosensing” (Grant no. 2011-0338) the projects PON01_02834 “Prometeo”, PONa3_00435 “Biomedpark@UMG” and FIRB “ReteNazionale di Ricerca sulle Nanoscienze ItalNanoNet” (cod. RBPR05JH2P_010, CUPB41J09000110005) financed from the Ministry of Education and Research, the project for Young researchers financed from the Ministry of Health “High Troughput analysis of cancer cells for therapy evaluation by microfluidic platforms integrating plasmonic nanodevices” (CUP J65C13001350001, project no. GR-2010-2311677) and Cancer biomarker detection using micro-structured/super-hydrophobic surfaces and advanced spectroscopy techniques (CUP J65C13001370001, project no. GR-2010-2320665) granted to the nanotechnology laboratory of the Department of Experimental Medicine of the University of Magna Graecia of Catanzaro.Notes and references
- S. Hanash, Disease proteomics, Nature, 2002, 422, 226–232 CrossRef PubMed.
- G. Simone, Proteomics, 2014, 14(9), 994–1000 CrossRef CAS PubMed.
- G. Simone, N. Malara, V. Trunzo, M. Renne, G. Perozziello, E. Di Fabrizio and A. Manz, Mol. BioSyst., 2014, 10(2), 258–265 RSC.
- G. Simone, P. Neuzil, G. Perozziello, N. Malara, M. Francardi, E. Di Fabrizio and A. Manz, Lab Chip, 2012, 12, 1500–1507 RSC.
- G. Simone and G. Perozziello, Micro Nanosyst., 2010, 2, 261–268 CrossRef CAS.
- G. Perozziello, J. Møllenbach, S. Laursen, E. Di Fabrizio, K. Gernaey and U. Krühne, Microelectron. Eng., 2012, 98, 655–658 CrossRef CAS.
- G. Perozziello, G. Simone, P. Candeloro, F. Gentile, N. Malara, R. La Rocca, M. Coluccio, S. A. Pullano, L. Tirinato, O. Geschke and E. Di Fabrizio, Micro Nanosyst., 2010, 2, 227–238 CrossRef CAS.
- F. Bundgaard, G. Perozziello and O. Geschke, J. Mech. Eng. Sci., 2006, 220(11), 1625–1632 Search PubMed.
- G. Simone and G. Perozziello, J. Nanosci. Nanotechnol., 2011, 11, 2057–2063 CrossRef CAS PubMed.
- G. Simone, G. Perozziello, G. Sardella, I. Disegna, S. Tori, N. Manaresi and G. Medoro, Microsyst. Technol., 2010, 16, 1269–1276 CrossRef CAS.
- G. Perozziello, F. Bundgaard and O. Geschke, Sens. Actuators, B, 2008, 130(2), 947–953 CrossRef CAS.
- A. Kiana, A. Fok, L. A. Sasso, N. Kamdar, Y. Guan, Q. Sun, A. Undarband and J. D. Zahn, Lab Chip, 2011, 11, 2858–2868 RSC.
- V. Kiermer, Nat. Methods, 2005, 2, 91 CrossRef CAS.
- A. B. Fuchs, A. Romani, D. Freida, G. Medoro, M. Abonnenc, L. Altomare, I. Chartier, D. Guergour, C. Villiers, P. N. Marche, M. Tartagni, R. Guerrieri, F. Chatelain and N. Manaresi, Lab Chip, 2006, 6, 121–126 RSC.
- D. J. Harrison, K. Fluri, K. Seiler, Z. Fan, C. S. Effenhauser and A. Manz, Science, 1993, 261, 895–897 CAS.
- http://www.miltenyibiotec.com.
- X. Chen and D. F. Cui, Information Storage And Processing Systems, Microsyst. Technol., 2009, 15, 667–676 CrossRef CAS.
- K. Lange and M. Rapp, Anal. Biochem., 2008, 377, 170–175 CrossRef PubMed.
- Q. G. Li and W. Ji, Analyst, 2010, 135, 441–451 CrossRef PubMed.
- L. A. Tessler, J. G. Reifenberger and R. D. Mitra, Anal. Chem., 2009, 81, 7141–7148 CrossRef CAS PubMed.
- S. A. Maier, Plasmonics: Fundamentals and Applications, Springer Verlag, 2007 Search PubMed.
- E. Hutter and J. H. Fendler, Adv. Mater., 2004, 16, 1685 CrossRef CAS.
- S. Ekgasit, C. Thammachareon and W. Knoll, Anal. Chem., 2004, 76, 561 CrossRef CAS PubMed.
- J. A. Schuller, E. S. Barnard, W. Cai, Y. C. Jun, J. S. White and M. L. Brongersma, Nat. Mater., 2010, 9, 193 CrossRef CAS PubMed.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102 CrossRef CAS PubMed.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392 CrossRef CAS PubMed.
- J. B. Goh, R. W. Loo, R. A. McAloney and M. C. Goh, Anal. Bioanal. Chem., 2001, 374, 54 CrossRef PubMed.
- S. A. Maier, P. G. Kik, H. A. Atwater, S. Meltzer, E. Harel, B. E. Koel and A. A. G. Requicha, Nat. Mater., 2003, 2, 229–232 CrossRef CAS PubMed.
- N. Fang, H. Lee, C. Sun and X. Zhang, Science, 2005, 308, 534 CrossRef CAS PubMed.
- L. Surbhi, S. Link and N. J. Halas, Nat. Photonics, 2007, 1, 641–648 CrossRef.
- D. A. Schultz, Curr. Opin. Biotechnol., 2003, 14, 13–22 CrossRef CAS PubMed.
- W. R. Premasiri, D. T. Moir, M. S. Klempner, N. Krieger, G. Jones II and L. D. Ziegler, J. Phys. Chem. B, 2005, 109, 312–320 CrossRef CAS PubMed.
- N. Shuming and S. R. Emory, Science, 1997, 275, 1102 CrossRef.
- L. Huang, S. J. Maerkl and O. J. F. Martin, Opt. Express, 2009, 8, 6018 CrossRef.
- E. Ouellet, C. Lausted, T. Lin, C. W. Yang, L. Hood and E. T. Lagally, Lab Chip, 2010, 10, 581 RSC.
- Y. Miki, J. Swensen, D. Shattuck-Eidens, P. A. Futreal, K. Harshman, S. Tavtigian, Q. Liu, C. Cochran, L. M. Bennett and W. Ding, Science, 1994, 266, 66–71 CAS.
- R. S. Williams and J. N. Glover, J. Biol. Chem., 2003, 278, 2630–2635 CrossRef CAS PubMed.
- R. S. Williams, D. I. Chasman, D. D. Hau, B. Hui, A. Y. Lau and J. N. Glover, J. Biol. Chem., 2003, 278, 53007–53016 CrossRef CAS PubMed.
- B. Quaresima, M. C. Faniello, F. Baudi, T. Crugliano, M. Di Sanzo, G. Cuda, F. Costanzo and S. Venuta, Oncol. Rep., 2006, 16, 811–815 CAS.
- F. Gentile, M. L. Coluccio, N. Coppedè, F. Mecarini, G. Das, C. Liberale, L. Tirinato, M. Leoncini, G. Perozziello, P. Candeloro, F. De Angelis and E. Di Fabrizio, ACS Appl. Mater. Interfaces, 2012, 4, 3213–3224 CAS.
- C. Krafft, T. Knetschke, R. H. W. Funk and R. Salzer, Vib. Spectrosc., 2005, 38, 85–93 CrossRef CAS.
- P. Candeloro, L. Tirinato, N. Malara, A. Fregola, E. Casals, V. Puntes, G. Perozziello, F. Gentile, M. L. Coluccio, G. Das, C. Liberale, F. De Angelis and E. Di Fabrizio, Analyst, 2011, 136, 4402–4408 RSC.
- G. Das, F. Mecarini, F. Gentile, F. De Angelis, H. G. M. Kumar, P. Candeloro, C. Liberale, G. Cuda and E. Di Fabrizio, Biosens. Bioelectron., 2009, 24(6), 1693–1699 CrossRef CAS PubMed.
- I. Harada, T. Miura and H. Takeuchi, Spectrochim. Acta, Part A, 1986, 42(2–3), 307–312 CrossRef.
| This journal is © The Royal Society of Chemistry 2014 |