
Synthesis, crystal structures and phase transformation of the new solid-state forms of tetrandrine†
Abstract
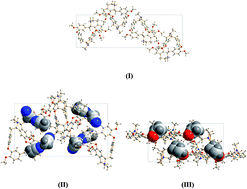
In this study, the solid-state forms and phase transformation of tetrandrine (Tet) were investigated and characterized using X-ray diffraction and thermal analysis. Crystal structures of form I, acetonitrile and acetone solvates were elucidated using single-crystal X-ray diffraction. Form I and the acetonitrile solvate were both determined to crystallize in the orthorhombic space group P212121 with Z′ = 1. The unit cell parameters were a = 7.12371 (10) Å, b = 11.92092 (16) Å, and c = 38.3027 (2) Å for form I; a = 9.70383 (10) Å, b = 15.06419 (15) Å, and c = 26.1648 (3) Å for the acetonitrile solvate, which present obvious differences. The acetone solvate was found to crystallize in the monoclinic space group P21 with Z′ = 2. The unit cell parameters were a = 18.7432 (5) Å, b = 9.9123 (3) Å, and c = 19.3410 (6) Å for the acetone solvate. Significant differences are observed among the host molecular skeletons of these three crystal structures from the overlay diagrams. The crystal voids and Hirshfeld surface 2D fingerprint plots were calculated for the first time. The solvatomorphic forms have better dissolution than form I.
 Please wait while we load your content...
Please wait while we load your content...

