
TiO2 nanotubes with tunable morphologies
Abstract
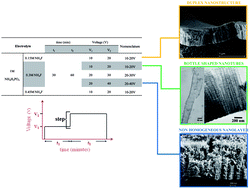
Titanium anodic oxide layers with a bottle shaped nanotubular structure have been grown in an electrolyte containing NH4F, applying voltage steps. The grown layers were analyzed using scanning and transmission electron microscopy (SEM and TEM), and Rutherford backscattering spectroscopy (RBS). The results show that a concentration of 0.15 M of NH4F in the anodizing bath, and a step of 10 to 20 V produces an oxide with a double morphology comprised of nanotubes at the oxide/metal interface and nanopores at the oxide/electrolyte interface of the anodic layer. Higher concentration of F− in the bath, 0.3 and 0.45 M NH4F, enhanced the chemical dissolution of the anodic layer resulting in nanotubular structures along the oxide layer. Therefore, the bottle shaped nanotubular structures that show a well defined morphology are obtained in a bath containing a concentration of 0.3 M NH4F and applying a voltage step of 10–20 V.
 Please wait while we load your content...
Please wait while we load your content...

