
Prediabetes: grounds of pitfall signalling alteration for cardiovascular disease
Sameer N. Goyal*a,
Shaikh Haideralia,
Navya Reddy Ma,
Dharamvir Singh Aryab and
Chandragouda R. Patila
aCardiovascular Pharmacology Division, Department of Pharmacology, R. C. Patel Institute of Pharmaceutical Education and Research, Shirpur, Dhule, Maharashtra, India. E-mail: goyal.aiims@gmail.com; Fax: +91 02563 251808; Tel: +91 02563 255 189, +91 955 291 6993
bDepartment of Pharmacology, All India Institute of Medical Sciences, New Delhi-110029, India
First published on 27th October 2014
Abstract
Impaired glucose metabolism either in prediabetes or diabetes mellitus is one of the detrimental root causes of premature mortality throughout the world. Uncontrolled prediabetes coincides with the induction of diabetic mellitus and associated cardiovascular diseases (CVDs). Needless to mention, impaired glucose metabolism, including impaired fasting glucose (IFG) and impaired glucose tolerance (IGT), have been known individually or in combination as the prediabetic stage but by itself it is not diabetes mellitus. Impaired β-cell function, insulin resistance, increased level of free fatty acids, hyperinsulinemia and down-regulation of GLUT-4 are critical impairments during prediabetes. The vascular endothelium sustains the free flow of blood in vessels by normalizing vascular tone by releasing numerous endothelial-derived factors. However, in recent studies a marked impairment in endothelial-derived factors has been observed in prediabetes. Thus, the impaired endothelial-derived factors could make prediabetic patients more vulnerable to cardiovascular disease pathology. Nobel laureates, Robert Furchgott, Louis Ignarro and Ferid Murad (1998) discovered a novel signalling molecule, nitric oxide (NO), identified as an endothelium-derived relaxing factor. This imperative mediator has potent vasodilatory, anti-platelet, anti-proliferative, and anti-inflammatory actions in vessels. Endothelium-derived NO generation is mediated through the activation of PI3-K-Akt-eNOS-NO signalling pathways. Therefore, conspicuous destruction in PI3-K-Akt-eNOS-NO signalling has been revealed in prediabetes and renders individuals more susceptible to CVDs. Several research reports have defined prediabetes as a platform for diabetes mellitus and associated CVDs. But the molecular alteration during prediabetes is unclear; however, the signalling modulator may be an imperative issue and may open a prerequisite new vista for novel research. In this review, we have critically discussed the possible signalling alteration in prediabetes.
1. Introduction
Impaired glucose metabolism is an overt sign of insulin dependent and non-dependent diabetes mellitus over a period of many years, which is a major culprit of mortality with towering prevalence in developed and developing countries.1 Prediabetes was first expressed by the World Health Organization in 1980 as impaired glucose tolerance.1 According to the American Diabetes Association, average blood glucose measurement levels in prediabetes for A1C test, fasting plasma glucose (FPG) test and oral glucose tolerance test (OGTT) are 5.7% to 6.4%, 100 mg dL−1 to 125 mg dL−1 and 140 mg dL−1 to 199 mg dL−1 respectively.2 However, a huge irony of fate is its awareness. The latest statistics suggest that 1 in 3 US adults have prediabetes, but only 11% are aware of their status.3 Generally, prediabetes is defined as the transitional metabolic state between normal and diabetic glucose homeostasis.4 Impaired glucose metabolism, including impaired fasting glucose (IFG) and impaired glucose tolerance (IGT) is referred to individually or in combination as the prediabetic stage but by itself is not diabetes.5 IFG predominantly concerns hepatic insulin resistance sensitivity, whereas muscle insulin resistance is responsible for IGT.6 Hepatic insulin resistance and unreliable insulin secretion in isolated IFG bring excessive production of fasting hepatic glucose creation, which instigates fasting hyperglycemia.7 Thus, diminishing of the early insulin response in combination with hepatic insulin resistance consequently increases the plasma glucose level. But the defence of late insulin secretion combined with normal muscle insulin sensitivity permits glucose levels to be restored to the primary value in isolated IFG.6 In contrast, hepatic insulin resistance and late insulin secretion become imperfect in isolated IGT, which results in prolonged hyperglycemia after glucose intake.4 Insulin resistance and impaired β-cell function are the crucial imperfections examined in diabetes mellitus, and both these defects are noticed in IGT and IFG.8 These important risk markers are major contributors of diabetes, retinopathy, neuropathy, vascular endothelial dysfunction and increased risk of cardiovascular pathology9 (Fig. 1).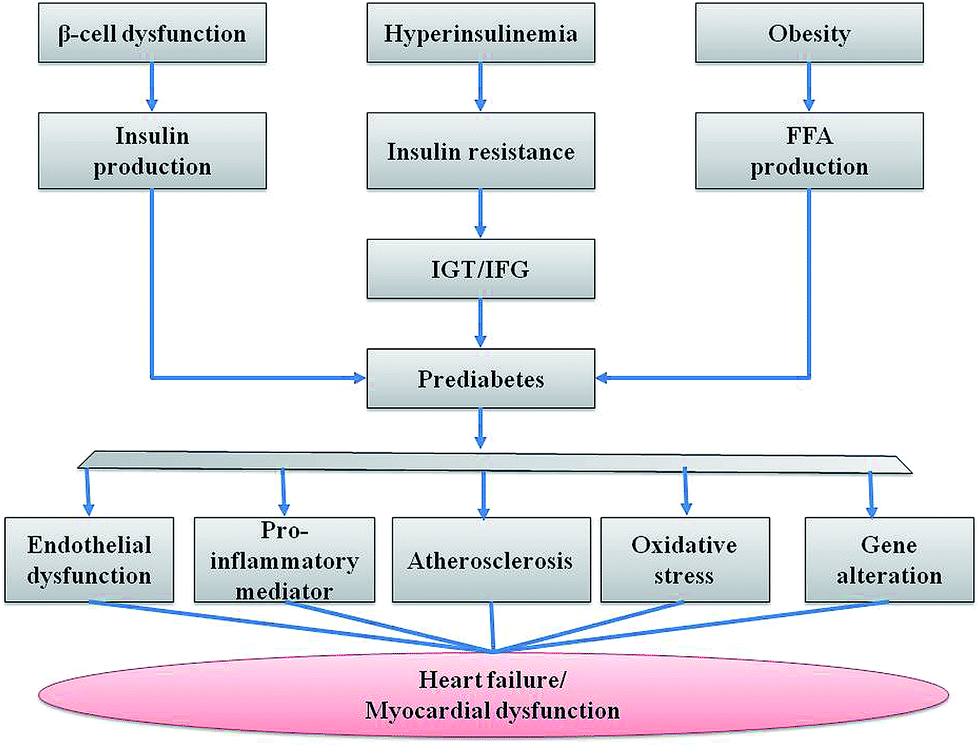 | ||
| Fig. 1 Prediabetic complication pathways leading to myocardial dysfunction. | ||
The endothelium is the innermost lining of blood vessels, principally involved in the regulation of vascular tone and maintenance of free flow of blood through vessels. C-type natriuretic peptide (CNP) and endothelin-1 (ET-1) are potent vasodilator and vaso-contracting endothelium derived factors which regulate vascular homeostasis. However, evidence suggests that the decreased level of endothelial-derived factor CNP, increased level of ET-1 and thickness of tunica intima are the key signalling alterations in IGT associated atherosclerosis and cardiovascular complications.10 In addition, phosphatidylinositide 3-kinase (PI3K) followed by protein kinase-B (PKB or Akt) signalling cascade is widely demonstrated for cardiac protection via activation of endothelial nitric oxide synthase (eNOS). This triggered eNOS to provoke the generation and bioavailability of the novel biosensor nitric oxide (NO), which has potent vasodilatory, anti-platelet, anti-proliferative, and anti-inflammatory actions in vessels.1 However, some recent findings suggest the alleviation of this innovative PI3K-Akt-eNOS-NO survival pathway during prediabetes conditions.11 Hypertriglyceridemia is a predisposed risk factor allied with atherosclerosis, vascular endothelial dysfunction (VED), and cardiovascular diseases (CVDs).12 In spite of this, increased levels of triacylglycerol, blood pressure and decreased activity of high density lipoprotein (HDL)-associated phospholipase A2 has been studied in prediabetic subjects.13 Moreover, reactive oxygen species (ROS) and tumor necrosis factor alpha (TNF-α) are key triggering molecules in the pathogenesis of endothelial dysfunction and associated cardiovascular disorders.14 However, ROS and TNF-α are markedly over diligent in prediabetes. In addition to TNF-α, increased plasma levels of granulocyte–monocyte colony-stimulating factor (GM-CSF0) interleukin-5 (IL-5), interleukin-6 (IL-6) and interleukin-7 (IL-7) have been registered in prediabetes.15 Thus, activated pro-inflammatory cytokine and dendritic cells (DCs) play a crucial role in the development of cardiovascular diseases.16 In spite of these signalling variations, sensitization of α-1 and α-2 adrenoreceptor signalling also has been perceived in prediabetes. Furthermore, this sensitization of α-1 and α-2 adrenoreceptor revealed the contribution of prediabetes to associated coronary vasoconstriction and cardiac ischemia.17 Prediabetes associated alteration in gene expression also asserted in the pancreatic islets resulting in further down-regulation of several genes participating in glucose metabolism and insulin signalling pathways in the next generation.18 Taken together, the evidence in terms of gradual decay in beta-cell function or increased insulin resistance and alteration in survival signalling pathways could impact significantly the action of drugs for treating cardiovascular complications associated with prediabetes. The present review delineates crucial pathophysiology and various signalling pathways identified to be involved in the pathogenesis of prediabetes.
2. Prevalence and pathophysiology
Prediabetes is the foremost trigger feature of diabetes mellitus and is a pivotal bench mark of VED and CVDs. Prevalence of prediabetes is rapidly growing in the population of developed and developing countries.19 The fact sheet of the National Health and Nutrition Examination Survey (NHANES) described 35% of people in the US aged ≥20 years and 50% of those ≥65 years suffered from prediabetes in 2005–2008, defined by the measurement of HbA1c concentrations. In estimations for 2010, 79 million adults were calculated from percentages of the entire US population.20 As per the 2011 report of the International Diabetes Federation (IDF), the prevalence of this insidious disease has been estimated to reach 472 million by the year 2030.4 Unreliable generation, bioavailability and sensitivity of insulin with respect to plasma glucose level leads to hyperglycaemia. The increased production of fasting hepatic glucose is carried out by virtue of hepatic insulin resistance and variable insulin secretion in isolated IFG, which instigates the fasting hyperglycemia.7 However, weaker early insulin response and hepatic insulin resistance consequently increase the plasma glucose level. In fact, this accumulated plasma level could be restored by late insulin secretion in isolated IFG.6 In contrast, the imperfection in hepatic insulin resistance and late insulin secretion in IGT results in prolonged hyperglycemia and prediabetes.4 Free fatty acids (FFAs) and cytokines released directly worsen insulin sensitivity. FFA provokes ROS and blunts the activation of insulin receptor substrate 1 (IRS-1). This alleviated IRS-1, consequently debilitating PI3K-Akt signalling and glucose transporter-4 (GLUT-4) in skeletal muscle and adipose tissue and therefore down-regulation of GLUT-4 leads to insulin resistance and diabetes mellitus.21 Obesity-linked insulin resistance could increase the level of cholesterol and that may be a core basis of prediabetes, diabetes mellitus and associated CVDs.2 Obesity also could promote an inflammatory pathway in pancreatic islets and lead to β-cell dysfunction. Thus, β-cell dysfunction may develop into prediabetes and diabetes.22 Hence β-cell damage leads to increased insulin release, leading to hyperinsulinemia. Hyperinsulinemia accelerates insulin resistance and induces a rise in circulating cholesterol.23 In this manner, the increased level of FFAs, insulin resistance, cholesterol, β-cell dysfunction, hyperinsulinemia and down-regulation of GLUT-4 are indeed pitfall causes of prediabetes onset.IFG and IGT are the precursors of type 2 diabetes and associated CVDs in individuals with prediabetes. Moderate to severe insulin resistance and impaired first and second phase insulin secretion occurs in the muscle of subjects with isolated IGT, whereas mild insulin resistance in liver, impaired first phase insulin secretion and almost normal muscle insulin sensitivity is seen in individuals with IFG.24 Apart from insulin resistance, excess fatty acids, adipokines etc. released from adipose tissue in individuals with prediabetes predisposes them to diabetes and associated cardiovascular diseases.9 These adipokines and glucose activate specific intracellular signaling pathways in the endothelium, resulting in endothelial dysfunction in diabetes.25 Epidemiological studies have shown that prediabetes confers an increased risk of cardiovascular disease. More or less 70% of individuals with prediabetes progress into a diabetic state with an additional risk for cardiovascular diseases as per an ADA expert panel.26 Prediabetes associated hyperglycemia mediated adverse effects through multiple pathways such as polyol, hexosamine, protein kinase C, and glycation pathways are the major factors contributing to cardiovascular risk by generating reactive oxygen species. Postprandial hyperglycemia negatively contributes to endothelial dysfunction, imparting higher CVD risk than fasting hyperglycemia by several mechanisms such as activation of NFkB and expression of monocytes, macrophages and smooth muscle cells.27 Moreover, Yuill reported the role of the β unit of transforming growth factor (TGFβ) in ventricular remodelling by activating the Akt–mTOR–p70S6K1 pathway and also by reactive oxygen species generation assisting further transforming of prediabetes into diabetes and cardiac dysfunction.28 Haemoglobin A1c levels between 5.7–6.4% in prediabetic patients are diagnostic markers of the future risk of diabetes and associated CVDs.27
3. Endothelium associated signalling alteration during prediabetes
The endothelium is an interior layer of the blood vessel that regulates numerous biological functions.29,24 Being the innermost layer, it regulates vascular tone and homeostasis by maintaining endothelium-derived relaxing (e.g., acetylcholine, ATP and ADP, substance P, bradykinin, histamine, thrombin, serotonin) and contracting factors (e.g., endothelin-1, arachidonic acid, Ca+ ionophore A23187, thromboxane A2, prostaglandin H2, and superoxide anions).30,9 Endothelium-derived hyperpolarizing factor CNP originates from the endothelium wall of the vessel and acts as an endothelial relaxing factor.31 Suppression of endothelial dependent vasorelaxation and expression of endothelial dependent vasocontraction leads to impairment in regulation of vascular homeostasis.32 CNP, an endothelium-derived hyperpolarizing factor, promotes cellular cGMP levels by binding to the G-protein receptor and natriuretic peptide receptor-B.33 CNP potently increases the level of IL-1β and TNF in response to lipopolysaccharides.34 Thus, a lower level of CNP is robustly correlated with VED, hypertension, atherosclerosis, thrombogenesis, re-stenosis and left ventricular diastolic dysfunction.35 In addition, increased levels of endothelin-1, a potent vasocontracting factor, also exaggerate VED and associated cardiovascular diseases.35 Regrettably, unsteadiness has been observed in both of these endothelial derived factors during prediabetes and insulin resistance, which is the pitfall hallmark of VED and associated cardiovascular disorders. Clinical evidence revealed the substantial reduction of CNP and considerably higher levels of endothelin-1 during prediabetes and thickness of endothelial layer, which addresses endothelial dysfunction6 (Fig. 2). This premise is supported by an imbalance of these endothelial derived factors in diabetes mellitus.36 Thus, the disparity in endothelial function during prediabetes because of impaired release of endothelium-derived relaxing and contracting factors may show the pathway of cardiovascular complications.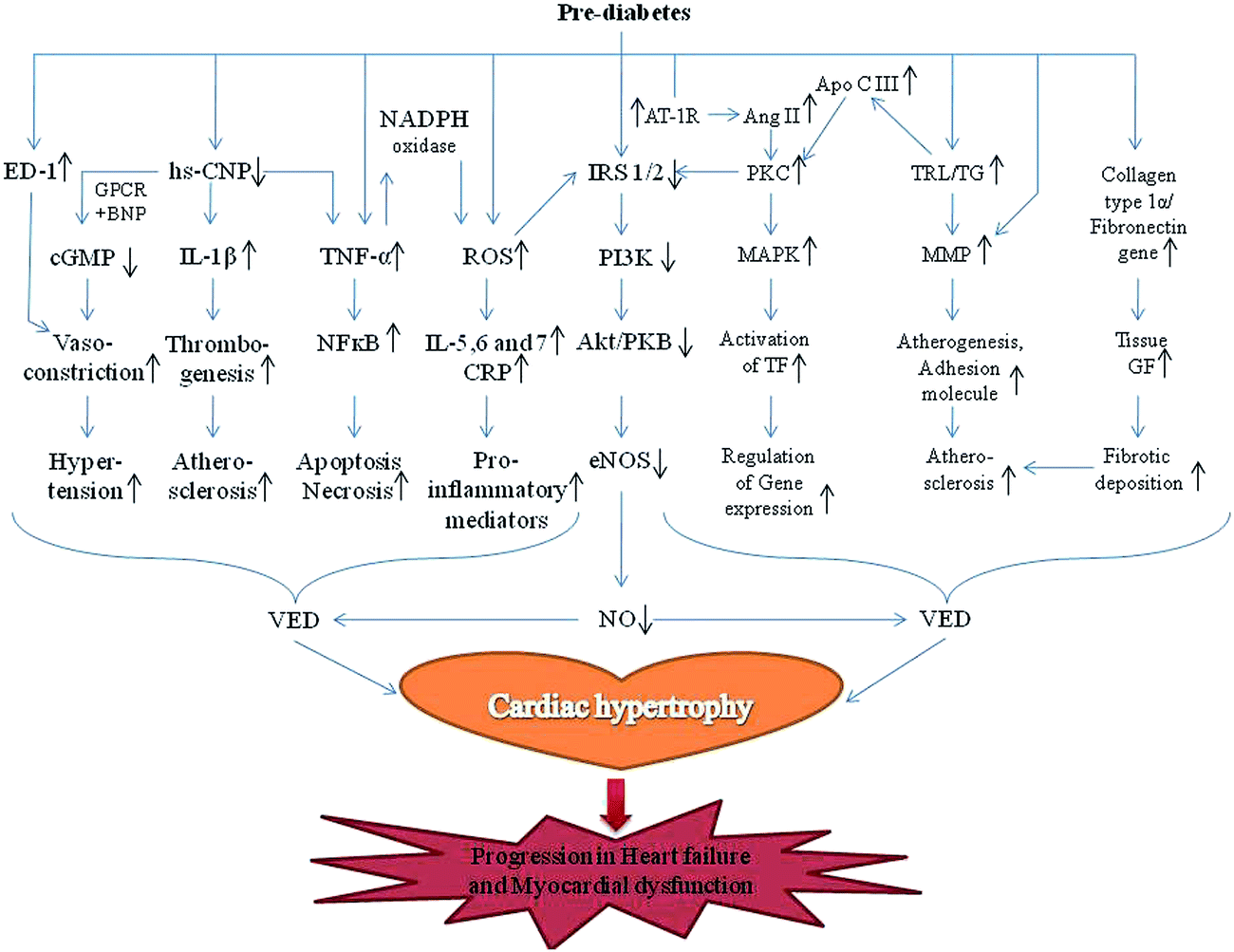 | ||
| Fig. 2 Prediabetic pathways involved in progression of heart failure. | ||
4. PI3K/PKB signalling alteration during prediabetes
PI3Ks, a family of kinases, are stimulated by tyrosine kinase receptors and G-protein coupled receptors. Akt/PKB is a family of serine/threonine kinase.37 PI3-K-Akt-eNOS-NO signalling has a pivotal role in the regulation of cardiovascular physiology.38 The PI3K/Akt signalling pathway is involved to invoke the stimulation and activation of eNOS, and associated NO formation in endothelial cells.39 The PI3K pathway also plays a vital role in the regulation of vascular endothelial function via activation of Akt/PKB, which consequently increases the phosphorylation of eNOS, and NO generation.40 Evidence suggested the regulating role of the PI3K-Akt signalling cascade in cardiomyocyte survival, angiogenesis, and inflammation.41 Several investigators have confirmed the fundamental role of PI3K/AKT signalling in cardiovascular protection. Prosopis glandulosa (honey mesquite tree), belonging to the Fabaceae (or legume) family, exerts cardio-protective and anti-hypertensive effects against prediabetes in a rat model via activation of PI3-K-Akt signalling pathways.42 Statins are potent inhibitors of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase, which is a rate-limiting enzyme for the synthesis of cholesterol, and are extensively used in the treatment of cardiovascular abnormalities including dyslipidemia, atherosclerosis and hypertension. This cardiovascular defensive action of statins highlights up-regulation of PI3-K-Akt-eNOS pro-survival pathways.43 Kobayashi et al. (2009) also imply the involvement of PI3-K-Akt associated eNOS production in the improved cardiac functioning and remodelling in cardioprotective effects of statins.44 Carbamylated erythropoietin, a carbamoyl derivative of erythropoietin, shows cardiovascular protective action by reducing myocardial apoptosis. Intriguingly, this attenuated cardiomyopathy by carbamylated erythropoietin involves stimulation of PI3K-Akt signaling pathways in rat myocardial cells.45A widely preferred clinical approach to inhibiting ischemia-induced cardiac apoptosis using Panax notoginseng saponins involves activation of PI3-K-Akt signaling pathways.46 Likewise, fibroblast growth factor 21, a hepatic metabolic regulator, exerts a cardiovascular protective action in lean and obese rat hearts in response to ischemia-induced injury through autocrine–paracrine pathways. Interestingly, fibroblast growth factor 21 associated cardioprotection against ischemia-induced injury is mediated through PI3K/Akt, ERK1/2 and AMPK pathways.47 Moreover, naturally occurring antioxidants, such as grape seed proanthocyanidin, defends cardiomyocytes against ischemia/reperfusion (I/R) injury by activation of Akt-eNOS signalling.48 SO2, which is believed to be a pollutant and toxic gas, exerts cardiovascular protection against myocardial I/R injury in rats. SO2 preconditioning significantly reduces myocardial infarct size and myocardial caspase-3 and 9 activities. This protective effect of SO2 preconditioning is suggested to involve the PI3K/Akt pathway.49 Ref. 45 reported that N-myc downstream-regulated gene 2 exerts a novel action of PI3K/Akt signaling in insulin mediating cardioprotection.50 Cuadrado et al. (2011) demonstrated that labdane diterpenes, which are well known for their cytoprotective and anti-inflammatory actions, also provide cardio-protection against anoxia-reperfusion-induced injury by anti-apoptosis action in an isolated heart by activating PI3K/AKT signalling.51 Similarly, Sun et al. (2011) have reported that the cardioprotective actions of tanshinone IIA also mediate through the Akt-dependent pathway in diabetic cardiomyopathy.52 Luteolin, a naturally occurring polyphenol flavonoid, exerts myocardial protection through the PI3K/Akt pathway in an experimental model of ischemia/reperfusion.53 β-Cadenin targeted remote ischemic preconditioning exerts potent myocardial protection by activating the PI3K/Akt signalling pathway.54 Activated protein C (APC) mediated cardioprotection against I/R injury also involves improvement of endothelial function via AKT1 activation.54 Kallistatin balances the vascular homeostasis by attenuating endothelial apoptosis through activation of the Akt-eNOS signaling pathway.55 The PI3-K-Akt associated signalling cascade plays an imperative role in cardiac function, including cardiac contractility.56 The PI3-K-Akt associated cardioprotective action against I/R injury offers unique opportunities for improving cardiac physiology.57 Taken together, PI3-K-Akt-eNOS signalling activation plays an integral role in maintaining cardiovascular function and preventing cardiovascular disease pathology.
However, studies revealed that PI3-K-Akt-eNOS signalling is substantially down-regulated in prediabetic conditions.1 Prediabetes associated metabolic changes including perturbed endothelial function, sub-clinical inflammation,58 changes in adipokines, expansion of atherogenic dyslipidemia, raised levels of free fatty acids (FFAs), and changes in thrombosis and fibrinolysis leading to cardiovascular abnormalities. IRS diminution has been observed in insulin resistance and β-cell dysfunction, which leads to PI3K-Akt/eNOS pathway suppression and consequently generates the platform for vascular endothelial dysfunction and associated cardiovascular diseases.21 A marked suppression in PI3-K-Akt-eNOS signalling in endothelial cells is noted in hyperglycemia.59 In addition, hyperglycemia-associated impairment in PI3-K-Akt signaling might promote endothelial cell proliferative dysfunction in diabetes. In the vascular system, endothelium-dependent relaxation is impaired in diabetes mellitus.59 Ref. 37 reported a significant down-regulation in PI3-K activity with a consequent decline in phosphorylation of downstream targets, including protein kinase B (PKB)/Akt, mTOR and FoxO in prediabetes associated obese groups.42 In addition, Hanson et al., (2010) demonstrated that insulin dependent phosphorylation of human erythrocyte cAMP is controlled by PI3K and PDE 3, which may lead to endothelial shrinkage.60 Moreover, Al-Suhaimi and Shehzad (2013) support the obstruction of the PI3K-dependent pathway in insulin resistance, associated with leptin deficiency.61 Since the PI3-K-Akt pathway regulates the function of vascular endothelium, its impaired activation in prediabetes might negatively influence the function of vascular endothelium, and might lead to cardiovascular disease pathology. Therefore, suppression of PI3-K-Akt-eNOS signalling might cause exaggerated cardiovascular disease pathology in patients with insulin resistance and prediabetes.
5. Hypertriglyceridemia associated signalling alteration during prediabetes
Triglyceride (TG), a central core of lipoprotein, is transported in plasma and regulates the endothelial function. In spite of optimal LDL-C reduction upon statin monotherapy, the risk of continuous atherogenic dyslipidemia remained high. This signifies the significant contribution of additional factor in atherosclerosis.62 Triglyceride-rich lipoproteins (TRLs), including VLDL, VLDL remnants and CM remnants, independently promote atherogenesis. Partial hydrolysis of TRLs produces remnant species in the presence of lipoprotein lipase.62 These remnant species uptake cholesterol esters through the action of cholesterol ester transfer protein and lead to accumulation of lipid and foam cell generation. This continuous accumulation of foam cells in artery walls causes fatty streaks and atherosclerosis.62 Both clinical and preclinical studies suggest that the activation of numerous proatherogenic responses such as augmented recruitment, attachment of monocyte- and macrophage-derived inflammatory proteins, including TNF-α, IL-1β, monocyte chemotactic protein-1, intercellular adhesion molecule-1 and matrix metalloproteinase-3 (MMP-3), increased expression of adhesion molecules and direct vascular cytotoxicity via lipolytic byproducts have been produced by TRLs and their remnants.63 In addition, TRLs also associate with apo C-III content, which leads to activation of PKC followed by down-regulation of the IRS/PI3K/PKB/NO signalling pathway.64 Therefore, this evidence proved that a high level of TG is an important biomarker involved in the promotion of several atherogenic lipoproteins.65 However, this central core of lipoprotein, TG, is elevated in prediabetes, which may produce the mentioned signalling alteration, atherosclerosis and associated cardiovascular diseases.66 Hypertriglyceridemia associated atherosclerosis is a widely documented predictor of cardiovascular disease by decreasing the lipoprotein concentration.676. TNF-α and ROS associated signalling alteration during prediabetes
Cytokine and TNF-α kick off the regulation of numerous cellular signals comprising NF-κ B, MAP kinase, necrosis and apoptosis. The pleiotropic action of TNF-α is to regulate the production of ROS by initiation of NADPH oxidase.68 The activation of TNF-α stimulates NADPH oxidase and augments the level of ROS. ROS are involved in a complex array of cellular cascades. Insulin resistance, β-cell dysfunction and obesity are the mediators of activation of TNF-α and ROS associated signalling alteration in prediabetic conditions.15 ROS may decrease insulin releasing substrate-1 (IRS-1), followed by down-regulation of PI3K-Akt/eNOS pathway, insulin resistance, hyperinsulinemia and GLUT-4 downregulation leading to vascular endothelial dysfunction and associated cardiovascular diseases. Moreover,21 TNF-α itself activates several pro-inflammatory cytokines by increasing GM-CSF, IL-5, IL-6, IL-7 and dendritic cells.15 Thus, an increase in the level of IL-6 and GM-CSF in prediabetes may be a sign of impending cardiovascular pathology. In addition, increased C-reactive protein (CRP), which is a bio-marker of inflammatory processes, may indicate endothelial dysfunction and cardiovascular events in diabetic conditions. More recently, Angelidi et al. (2014) reported CRP as a potential novel marker of vascular integrity, atherosclerosis and cardiovascular risk.69 Eventually scientific data suggested CRP to be an individual future risk predictor for cardiovascular diseases in healthy individuals and patients with coronary syndrome. This pivotal evidence clarify the fact that CRP activation and its presence in plasma may indicate the existence of cardiovascular diseases. An increased level of CRP has been observed in prediabetes, which supports the presence of endothelial and cardiovascular dysfunction. This cardiovascular abnormality associated with an inflammatory pathway may be mediated by prediabetes signalling alteration by pro-inflammatory cytokines. Hence, these pro-inflammatory cytokines and ROS activated signalling alteration cascades may be critical root causes for cardiovascular disorder in prediabetic condition.7. Gene expression alteration during prediabetes
As per the findings of several research reports, hyperglycemia and increased activity of TGFβ1 is observed in prediabetes. D’Souza et al. (2011) evidenced that both hyperglycemia and increased activity of TGFβ1 regulate gene expression of the extracellular matrix (ECM). Moreover, the authors also revealed the increased expression of collagen type 1α and fibronectin by measuring increased fibrotic deposition in GK rats (prediabetic Goto–Kakizaki rats).70 Thus, the increased level of the ECM regulator gene leads to the expression of connective tissue growth factor by fibroblast differentiation. Matrix metalloproteinases (MMP), a type of endopeptidase, are involved in the development of plaque, atherosclerosis and cardiovascular pathology.71 In addition, MMP-2 and MMP-9 are significantly upregulated in genetically modified prediabetic rats.70 Furthermore, it has been disclosed that prediabetes could be inherited transgenerationally by human germ cells. Wei and his coworkers have demonstrated that the offspring of prediabetic fathers show altered gene expression in the pancreatic islets. Prediabetic paternal cytosine methylation has been observed as a significant alteration in their offspring epigenomic profile. These alterations produce reproducible changes in methylation of the consequent insulin signalling gene. The methylome sequence of spermetazoa is distorted in prediabetic paternal patients and that causes a malformed insulin signalling gene in the pancreatic islets of offspring.72 Thus, down-regulation of glucose metabolism and insulin signalling regulator gene has been observed in offspring of prediabetic parents. Hence, these observations uniquely demonstrate that prediabetes associated gene alteration leads to serious changes such as insulin signalling, glucose metabolism, increased fibrotic deposition and the development of plaque and atherosclerosis.8. Miscellaneous signalling alteration during prediabetes
Apart from the above discussed signalling pathways, several other pathways are also involved. Many laboratories demonstrated the role of the sympathetic α-adrenergic receptor in regulating coronary blood circulation.73 Baumgart et al. (1999) suggested that the reduction in coronary blood flow occurs through α-1 and α-2 adrenoreceptor activation induced vasoconstriction.74 Furthermore, the authors suggest that augmented microvascular constriction occurs through epicardial α-1 and α-2 adrenergic receptor activation, which is associated with atherosclerosis and can cause myocardial ischemia.75 Recent extensive evidence is in agreement with the improvement in coronary blood flow, myocardial function and metabolism by blockade of α-1 and α-2 adrenoceptors. A larger contribution of α-2 adrenoceptor than α-1 in the constriction of the resistive microcirculation is also experimentally substantiated.76 This existing evidence reveals the impact of α-1 and α-2 adrenergic receptors in microvascular constriction and associated cardiovascular diseases. Unluckily, prediabetes is associated with sensitization of α-1 and α-2 adrenergic receptor signalling, which could reduce control of coronary blood flow.77 Augmented levels of Ang II, a peptide hormone, critically leads to congestive heart failure related morbidity and mortality.78 Ang II considerably activates the angiotensin-II receptor, type 1 (AT-1R) and leads to cardiac hypertrophy by activating various transcription pathways.79 Zhang et al. (2005) observed the increased level of Ang II during prediabetic metabolic syndrome, which can significantly increase coronary vasoconstriction both in vitro (isolated coronary arterioles, 60–110 μm) and in vivo (anesthetized open-chest dog).80 Increased concentration of leptin in obese individuals associated with prediabetes is an independent risk factor for attenuating coronary dilation, which results in coronary vascular diseases and VED.819. Therapeutic implications of prediabetes
Adopting a balanced diet and non-sedentary lifestyle, such as exercise in daily routine, contributes to reducing haemoglobin A1c levels rapidly. Medications such as anti-cholestrol, anti-lipase and GLP-1 analogues are used to control obesity, which is referred as a major risk factor in prediabetes.27 Metformin and acarbose treatment, which have been implicated in 1st line prediabetic therapy over the last four decades, offer promising protection.82 Apart from the above drugs, DPP-4 inhibitors, insulin and insulin secretagogues such as sulfonylureas, incretins and its analogues offer β-cell protection and improve insulin sensitivity through cellular growth and regeneration.83 NF kappa-B mediated inflammation leads to insulin sensitivity, atherogenecity and increased C-reactive protein concentrations, hence pharmacological targets of NF kappa-B such as aspirin seem to benefit prediabetic subjects. PPAR-γ receptor activators such as thiazolidinediones (TZD) have shown beneficial effects by increasing the expression of glucoregulatory molecules and enhanced insulin sensitivity.84 Chromium elicits therapeutic benefits in patients with insulin resistance by de-phosphorylation of protein tyrosine phosphatise, which is a negative regulator of the insulin receptor.85 Even though such medications exist, there are drawbacks such as high cost of medications, patient compliance and most of all the glycemic rebound that occurs immediately after cessation of medication. Hence, there is a need for novel pharmacological targets that can bias these drawbacks and can be used as a first line therapy in prediabetes as well as diabetes. Recent studies have revealed that overexpression of miRNA29/a/b/c increases diabetic manifestations and apoptosis in islets of NOD mice.86 This data may lead to a new direction of research in this field. Likewise, decreased NO bioavailability occurs due to eNOS dysfunction by the enzyme arginase. Hence, inhibitors of arginase and activators of eNOS may be of use in preventing endothelial dysfunction.10. Conclusions
It is well known fact that prediabetes plays a key role in pathogenesis of diabetes mellitus and CVD by altering several signalling pathways. Hence, there is an escalated interest in finding the most promising therapeutically targetable signalling cascade involved in CVD in prediabetes patients. In fact, recent bench and clinical studies have suggested that physical exercise and some therapeutic agents may have protective role in prediabetes. The involvement of molecular mechanisms and downstream effectors that provoke the development of cardiovascular risk have remained largely unclear. Moreover, a multipronged intervention is always the most effectual in exploiting new molecular entities. Thus, the present review throws light on the signalling alterations associated with prediabetes generated CVDs. Several studies have shown that the prediabetic heart is associated with alteration or down-regulation of CNP, ET-1, PI3-K-AKt-eNOS, TG, TNF-α, ROS, α-1 and α-2 adrenoreceptor, leptin and Ang II pathways. However, there is a need to meticulously trace out the targetable signalling perturbations in the metabolic disorders associated with the attenuation of cardioprotection during prediabetes. A paradigm shift in the generation of novel therapeutic approaches to prevent the pathogenesis of CVDs that can then be translated to clinical scenarioes in the near future is warranted.Conflict of interest
The authors declare that there are no conflicts of interest.Search methodology
Database searches using Google Scholar, Pubmed, and Science Direct were conducted up to 21st July 2014 to include up-to-date documented information in the present review article. The search was limited to English language papers. For data mining, the following MESH words were used in the databases mentioned above: prediabetes, IGT, IFG, signalling mechanism, PI3K/AKT signalling, eNOS, diabetes, myocardial infarction, apoptosis, reactive oxidative stress, caspase, antiapoptotic, OGTT, iIGT. In almost all cases, the original articles were obtained and the relevant data was extracted.Acknowledgements
The authors gratefully acknowledge the financial support received under Young Scientist Research Scheme (File no. SB/YS/LS-114/2013) of Science and Engineering Research Board (SERB), Department of Science and Technology, New Delhi, India.References
- A. K. Sharma and D. Khanna, Diabetes, Cell. Signalling, 2013, 25, 1149–1155 CrossRef CAS PubMed.
- http://www.diabetes.org/diabetes-basics/diagnosis/, accessed on April 10, 2014.
- Centers for Disease Control and Prevention (CDC), Awareness of prediabetes–United states, 2005–2010, Morbidity and Mortality Weekly Report, 2013, 62(11), 209–212 Search PubMed.
- A. G. Tabak, C. Herder, W. Rathmann, E. J. Brunner and M. Kivimäki, Lancet, 2012, 379, 2279–2290 CrossRef.
- D. M. Nathan, M. B. Davidson, R. A. DeFronzo, R. J. Heine, R. R. Henry, R. Pratley and B. Zinman, Diabetes Care, 2007, 30, 7539 Search PubMed.
- M. Kanat, L. Norton, D. Winnier, C. Jenkinson, R. A. DeFronzo and M. A. Abdul-Ghani, Acta Diabetol., 2011, 48, 209–217 CrossRef CAS PubMed.
- B. Göke, Diabetes Res. Clin. Pract., 1998, 40, 15–20 CrossRef.
- X. Y. Han, L. N. Ji and X. H. Zhou, Zhonghua Yixue Zazhi, 2004, 84, 1777–1780 CAS.
- S. M. Grundy, J. Am. Coll. Cardiol., 2012, 59(7), 635–643 CrossRef CAS PubMed.
- Y. Liu, J. Li, Z. Zhang, Y. Tang, Z. Chen and Z. Wang, Exp. Ther. Med., 2014, 7(3), 697–702 CAS.
- B. Huisamen, C. George, D. Dietrich and S. Genade, Cardiovasc. J. South Af., 2013, 24(2), 10–16 CrossRef CAS PubMed.
- M. Miller, N. J. Stone, C. Ballantyne, V. Bittner, M. H. Criqui, H. N. Ginsberg, A. C. Goldberg, W. J. Howard, M. S. Jacobson, P. M. Kris-Etherton, T. A. Lennie, M. Levi, T. Mazzone and S. Pennathur, Circulation, 2011, 123(20), 2292–2333 CrossRef PubMed.
- M. Hausding, K. Jurk, S. Daub, S. Kröller-Schön, J. Stein, M. Schwenk, M. Oelze, Y. Mikhed, J. G. Kerahrodi, S. Kossmann, T. Jansen, E. Schulz, P. Wenzel, A. B. Reske-Kunz, C. Becker, T. Münzel, S. Grabbe and A. Daiber, Basic Res. Cardiol., 2013, 108(6), 386 CrossRef PubMed.
- A. Magenta, S. Greco, C. Gaetano and F. Martelli, Int. J. Mol. Sci., 2013, 14(9), 17319–17346 CrossRef PubMed.
- R. Lucas, S. J. Parikh, S. Sridhar, D. H. Guo, J. Bhagatwala, Y. Dong, R. Caldwell, A. Mellor, W. Caldwell, H. Zhu and Y. Dong, Cytokines, 2013, 64(1), 310–315 CrossRef CAS PubMed.
- M. Hausding, K. Jurk, S. Daub, S. Kröller-Schön, J. Stein, M. Schwenk, M. Oelze, Y. Mikhed, J. G. Kerahrodi, S. Kossmann, T. Jansen, E. Schulz, P. Wenzel, A. B. Reske-Kunz, C. Becker, T. Münzel, S. Grabbe and A. Daiber, Basic Res. Cardiol., 2013, 108(6), 386 CrossRef PubMed.
- U. D. Dincer, A. G. Araiza, J. D. Knudson, P. E. Molina and J. D. Tune, Microcirculation, 2006, 13(7), 587–595 CrossRef CAS PubMed.
- Y. Wei, C. R. Yang, Y. P. Wei, Z. A. Zhao, Y. Hou, H. Schatten and Q. Y. Sun, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(5), 1873–1878 CrossRef CAS PubMed.
- T. D. Filippatos, E. C. Rizos, V. Tsimihodimos, I. F. Gazi, A. D. Tselepis and M. S. Elisaf, Lipids, 2013, 48(6), 547–555 CrossRef CAS PubMed.
- Centers for Disease Control and Prevention, National Diabetes Fact Sheet: national estimates and general information on diabetes and prediabetes in the United States, US Department of Health and Human Services, Centers forDisease Control and Prevention, Atlanta, GA, 2011 Search PubMed.
- A. R. Saltiel and C. R. Kahn, Nature, 2001, 414(6865), 799–806 CrossRef CAS PubMed.
- M. Chentouf, G. Dubois, C. Jahannaut, F. Castex, A. D. Lajoix, R. Gross and S. Peraldi-Roux, PLoS One, 2011, 6(8), e22–954 Search PubMed.
- E. D. Abel, K. M. O’Shea and R. Ramasamy, Arterioscler., Thromb., Vasc. Biol., 2012, 32(9), 2068–2076 CrossRef CAS PubMed.
- M. A. A. Ghani and R. A. Defronzo, Curr. Diabetes Rep., 2009, 9, 193–199 CrossRef PubMed.
- E. C. Eringa, E. H. Serne, R. I. Meijer, C. G. Schalkwijk, A. J. H. M. Houben, C. D. A. Stehouwer, Y. M. Smulders and V. W. M. van Hinsbergh, Rev. Endocr. Metab. Disord., 2013, 14, 39–48 CrossRef PubMed.
- http://www.thelancet.com, vol. 379, June 16, 2012, accessed on 12-10-2014.
- E. A. Nyenwe, T. W. Jerkins, G. E. Umpierrez and A. E. Kitabchi, Metabolism, 2011, 60(1), 1–23 CrossRef CAS PubMed.
- K. H. Yuill, Exp. Physiol., 2011, 829–830 Search PubMed.
- P. Balakumar, T. Kaur and M. Singh, Toxicology, 2008, 245(1–2), 49–64 CrossRef CAS PubMed.
- R. F. Furchgott and P. M. Vanhoutte, FASEB J., 1989, 3(9), 2007–2018 CAS.
- S. L. Sandow and M. Tare, Trends Pharmacol. Sci., 2007, 28(2), 61–67 CrossRef CAS PubMed.
- M. J. Morgan and Z. G. Liu, Mol. Cells, 2010, 30(1), 1–12 CrossRef CAS PubMed.
- R. S. Del, Peptides, 2013, 40, 93–98 CrossRef PubMed.
- S. Verma, Can. J. Cardiol., 2004, 20(suppl. B), 29B–31B Search PubMed.
- M. Iglarz and M. Clozel, J. Cardiovasc. Pharmacol., 2007, 50(6), 621–628 CrossRef CAS PubMed.
- T. Walther, H. P. Schuitheiss and C. Tschöpe, Cardiovasc. Res., 2001, 51(3), 562–566 CrossRef CAS.
- S. Masure, B. Haefner, J. J. Wesselink, E. Hoefnagel, E. Mortier, P. Verhasselt, A. Tuytelaars, R. Gordon and A. Richardson, Eur. J. Biochem., 1999, 265, 353–360 CrossRef CAS.
- Y. Fujio, T. Nguyen, D. Wencker, R. N. Kitsis and K. Walsh, Circulation, 2000, 101, 660–667 CrossRef CAS.
- J. X. Chen and B. Meyrick, Lab. Invest., 2004, 84, 182–190 CrossRef CAS PubMed.
- F. Gao, E. Gao, T. L. Yue, E. H. Ohlstein, B. L. Lopez, T. A. Christopher and X. L. Ma, Circulation, 2002, 105, 1497–1502 CrossRef CAS.
- B. H. Jiang and L. Z. Liu, Adv. Cancer Res., 2009, 102, 19–65 CrossRef CAS.
- B. Huisamen, C. George, D. Dietrich and S. Genade, Cardiovasc. J. South Af., 2013, 24, 10–16 CrossRef CAS PubMed.
- R. M. Bell and D. M. Yellon, J. Am. Coll. Cardiol., 2003, 41, 508–515 CrossRef CAS.
- N. Kobayashi, H. Takeshima, H. Fukushima, W. Koguchi, Y. Mamada, H. Hirata, Y. Machida, M. Shinoda, N. Suzuki, F. Yokotsuka, K. Tabei and H. Matsuoka, Am. J. Hypertens., 2009, 22, 176–182 CrossRef CAS PubMed.
- H. He, X. Qiao and S. Wu, Exp. Ther. Med., 2013, 6, 567–573 CAS.
- S. Chen, J. Liu, X. Liu, Y. Fu, M. Zhang, Q. Lin, J. Zhu, L. Mai, Z. Shan, X. Yu, M. Yang and S. Lin, J. Ethnopharmacol., 2011, 137, 263–270 CrossRef CAS PubMed.
- V. Patel, R. Adya, J. Chen, M. Ramanjaneya, M. F. Bari, S. K. Bhudia, E. W. Hillhouse, B. Tan, S. Harpal and M. Randeva, PLoS One, 2014, 9, 1–11 Search PubMed.
- Z. H. Shao, K. R. Wojcik, A. Dossumbekova, C. Hsu, S. R. Mehendale, C. Q. Li, Y. Qin, W. W. Sharp, W. T. Chang, K. J. Hamann, C. S. Yuan and T. L. Hoek, J. Cell. Biochem., 2009, 107, 697–705 CrossRef CAS PubMed.
- M. M. Zhao, J. Y. Yang, X. B. Wang, C. S. Tang, J. B. Du and H. F. Jin, Acta Pharmacol. Sin., 2013, 34, 501–506 CrossRef CAS PubMed.
- Z. Sun, G. Tong, N. Ma, J. Li, X. Li, S. Li, J. Zhou, L. Xiong, F. Cao, L. Yao, H. Wang and L. Shen, Basic Res. Cardiol., 2013, 108, 341 CrossRef PubMed.
- I. Cuadrado, M. Fernández-Velasco, L. Boscá and B. de Las Heras, Cell Death Dis., 2011, 2, 229 CrossRef PubMed.
- D. Sun, M. Shen, J. Li, L. Weijie, Y. Zhang, L. Zhao, Z. Zhang, Y. Yuan, H. Wang and F. Cao, Cardiovasc. Diabetol., 2011, 10, 4 CrossRef CAS PubMed.
- F. Fang, D. Li, H. Pan, D. Chen, L. Qi, R. Zhang and H. Sun, Pharmacology, 2011, 88, 149–158 CrossRef CAS PubMed.
- Y. Maehata, S. Miyagawa and Y. Sawa, PLoS One, 2012, 7, 38738 Search PubMed.
- B. Shen, L. Gao, Y. T. Hsu, G. Bledsoe, M. Hagiwara, L. Chao and J. Chao, Am. J. Physiol.: Heart Circ. Physiol., 2010, 299, 1419–1427 CrossRef PubMed.
- I. Kehat and J. D. Molkentin, Circulation, 2010, 122, 2727–2735 CrossRef PubMed.
- A. Dhanasekaran, S. K. Gruenloh, J. N. Buonaccorsi, R. Zhang, G. J. Gross, J. R. Falck, P. K. Patel, E. R. Jacobs and M. Medhora, Am. J. Physiol.: Heart Circ. Physiol., 2008, 294, 724–735 CrossRef PubMed.
- A. Festa, A. J. Hanley, R. P. Tracy, R. D’Agostino Jr and S. M. Haffner, Circulation, 2003, 108(15), 1822–1830 CrossRef CAS PubMed.
- H. O. Steinberg, M. Tarshoby, R. Monestel, G. Hook, J. Cronin, A. Johnson, B. Bayazeed and A. D. Baron, J. Clin. Invest., 1997, 100(5), 1230–1239 CrossRef CAS PubMed.
- M. S. Hanson, A. H. Stephenson, E. A. Bowles and R. S. Sprague, Exp. Biol. Med., 2010, 235(2), 256–262 CrossRef CAS PubMed.
- E. A. Al-Suhaimi and A. Shehzad, Eur. J. Med. Res., 2013, 18, 12 CrossRef CAS PubMed.
- B. G. Talayero and F. M. Sacks, Curr. Cardiol. Rep., 2011, 13(6), 544–552 CrossRef PubMed.
- S. Patel, R. Puranik, S. Nakhla, P. Lundman, R. Stocker, X. S. Wang, G. Lambert, K. A. Rye, P. J. Barter, S. J. Nicholls and D. S. Celermajer, Atherosclerosis, 2009, 204(2), 424–428 CrossRef CAS PubMed.
- A. Kawakami, M. Osaka, M. Tani, H. Azuma, F. M. Sacks, K. Shimokado and M. Yoshida, Circulation, 2008, 118(7), 731–742 CrossRef CAS PubMed.
- H. A. Hadi, C. S. Carr and J. Al Suwaidi, Vasc. Health Risk Manage., 2005, 1(3), 183–198 CAS.
- H. A. Al-Aubaidy and H. F. Jelinek, Redox Rep., 2014,(2), 87–91 CrossRef CAS PubMed.
- L. Perreault, B. C. Bergman, D. M. Hunerdosse, M. C. Playdon and R. H. Eckel, Obesity, 2010, 8, 1524–1531 CrossRef PubMed.
- M. J. Morgan and Z. G. Liu, Mol. Cells, 2010, 30(1), 1–12 CrossRef CAS PubMed.
- A. Angelidi, A. Melidonis, I. Protopsaltis, T. N. Sergentanis, A. Gritzapis, I. Kornezos, T. Economopoulos, G. Dimitriadis, E. Boutati and S. A. Raptis, Hormones, 2014, 13(2), 244–251 Search PubMed.
- A. D’Souza, F. C. Howarth, J. Yanni, H. Dobryznski, M. R. Boyett, E. Adeghate, K. R. Bidasee and J. Singh, Exp. Physiol., 2011, 96(9), 875–888 Search PubMed.
- I. M. Loftus, A. R. Naylor, P. R. Bell and M. M. Thompson, Br. J. Surg., 2002, 89(6), 680–694 CrossRef PubMed.
- Y. Wei, C. R. Yang, Y. P. Wei, Z. A. Zhao, Y. Hou, H. Schatten and Q. Y. Sun, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(5), 1873–1878 CrossRef CAS PubMed.
- R. M. Berne, H. Degeest and M. N. levy, Am. J. Physiol., 1965, 208, 763–769 CAS.
- D. Baumgart, C. Naber, M. Haude, O. Oldenburg, R. Erbel, G. Heusch and W. Siffert, Circ. Res., 1999, 85(10), 965–969 CrossRef CAS.
- D. Baumgart, M. Haude, G. Görge, F. Liu, J. Ge, G. Claudia, R. Erbel and G. Heusch, Circulation, 1999, 99(16), 2090–2097 CrossRef CAS.
- G. Heusch, J. Mol. Cell. Cardiol., 2011,(1), 16–23 CrossRef CAS PubMed.
- U. D. Dincer, A. G. Araiza, J. D. Knudson, P. E. Molina and J. D. Tune, Microcirculation, 2006, 13(7), 587–595 CrossRef CAS PubMed.
- P. Balakumar and G. Jagadeesh, J. Pharmacol. Res., 2010, 62(5), 365–383 CrossRef CAS PubMed.
- R. M. Califf and J. N. Cohn, Am. Heart J., 2000, 139(1 Pt 2), S15–S22 CrossRef CAS.
- C. Zhang, J. D. Knudson, S. Setty, A. Araiza, U. D. Dincer, L. Kuo and J. D. Tune, Am. J. Physiol.: Heart Circ. Physiol., 2005, 288(5), H2154–H2162 CrossRef CAS PubMed.
- J. D. Knudson, U. D. Dincer, G. M. Dick, H. Shibata, R. Akahane, M. Saito and J. D. Tune, Am. J. Physiol.: Heart Circ. Physiol., 2005, 289(3), H1038–H1046 CrossRef CAS PubMed.
- L. B. A. Rojas and M. B. Gomes, Diabetol. Metab. Syndr., 2013, 5–6 Search PubMed.
- O. R. Pour and S. D. Jack, Clin. Chem., 2011, 57(2), 215–220 CAS.
- J. M. Olefsky, J. Clin. Invest., 2000, 106 Search PubMed.
- J. D. Nicholas and T. Hunter, J. Pharm. Sci. Innovation, 2014, 298–305 Search PubMed.
- E. Roggli, S. Gattesco, D. Caille, C. Briet, C. Boitard, P. Meda and R. Regazzi, Diabetes, 2012, 61, 1742–1751 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |