DOI:
10.1039/C4RA10365C
(Communication)
RSC Adv., 2014,
4, 54144-54148
Xylariterpenoids A–D, four new sesquiterpenoids from the Xylariaceae fungus†
Received
13th September 2014
, Accepted 16th October 2014
First published on 17th October 2014
Abstract
Four new sesquiterpenoids, sesquiterpenoids A–D (1–4), were isolated from solid cultures of the Xylariaceae fungus (no. 63-19-7-3). Their structures were determined through NMR analyses, CD calculation, the in situ dimolybdenum CD method, the modified Mosher's method, and X-ray data analysis. The cytotoxicities of all compounds against HL-60, SMMC-7721, A-549, MCF-7 and SW480 human cancer cell lines were assayed.
Introduction
The Xylariaceae is one of the largest families of ascomycetes (over 60 genera and far more than 1000 species). Moreover, in excess of 500 metabolites have been reported from Xylariaceae fungi.1 Previous chemical investigations have revealed that this family produces a variety of secondary metabolites including dihydroisocoumarins, punctaporonins, cytochalasins, butyrolactones and succinic acid derivatives.2 During our ongoing search for bioactive secondary metabolites from endolichenic fungi,3 the chemical investigation of the Xylariaceae fungus (no. 63-19-7-3) was carried out, which led to the isolation of four new sesquiterpenoids (1–4). The planar structures of 1–4 were established primarily by NMR experiments. The absolute configurations of 1 and 2 were determined by CD and in situ dimolybdenum CD methods, while the absolute configurations of 3 and 4 were deduced via the modified Mosher's method and X-ray data analysis, respectively. Detail of the isolation, structural and absolute configurations elucidation, and cytotoxicity of all the compounds are reported herein.
Materials and methods
The fermented material of the Xylariaceae fungus (no. 63-19-7-3) was extracted with EtOAc, and the organic solvent was evaporated under vacuum to afford the dry crude extract (28.1 g). Then the crude extract was dissolved in 90% (v/v) aqueous MeOH (500 mL) and partitioned against the same volume cyclohexane to afford cyclohexane fraction (C, 15.3 g) and aqueous MeOH fraction (W, 12.5 g). The aqueous MeOH fraction was successively subjected to column chromatography over ODS, Sephadex LH-20 and reversed-phase HPLC (RPHPLC) to obtain compounds 1–4. Then, the cytotoxicity assay of compounds 1–4 was performed. The detailed experimental procedures are available in the ESI.†
Results
Structural elucidation
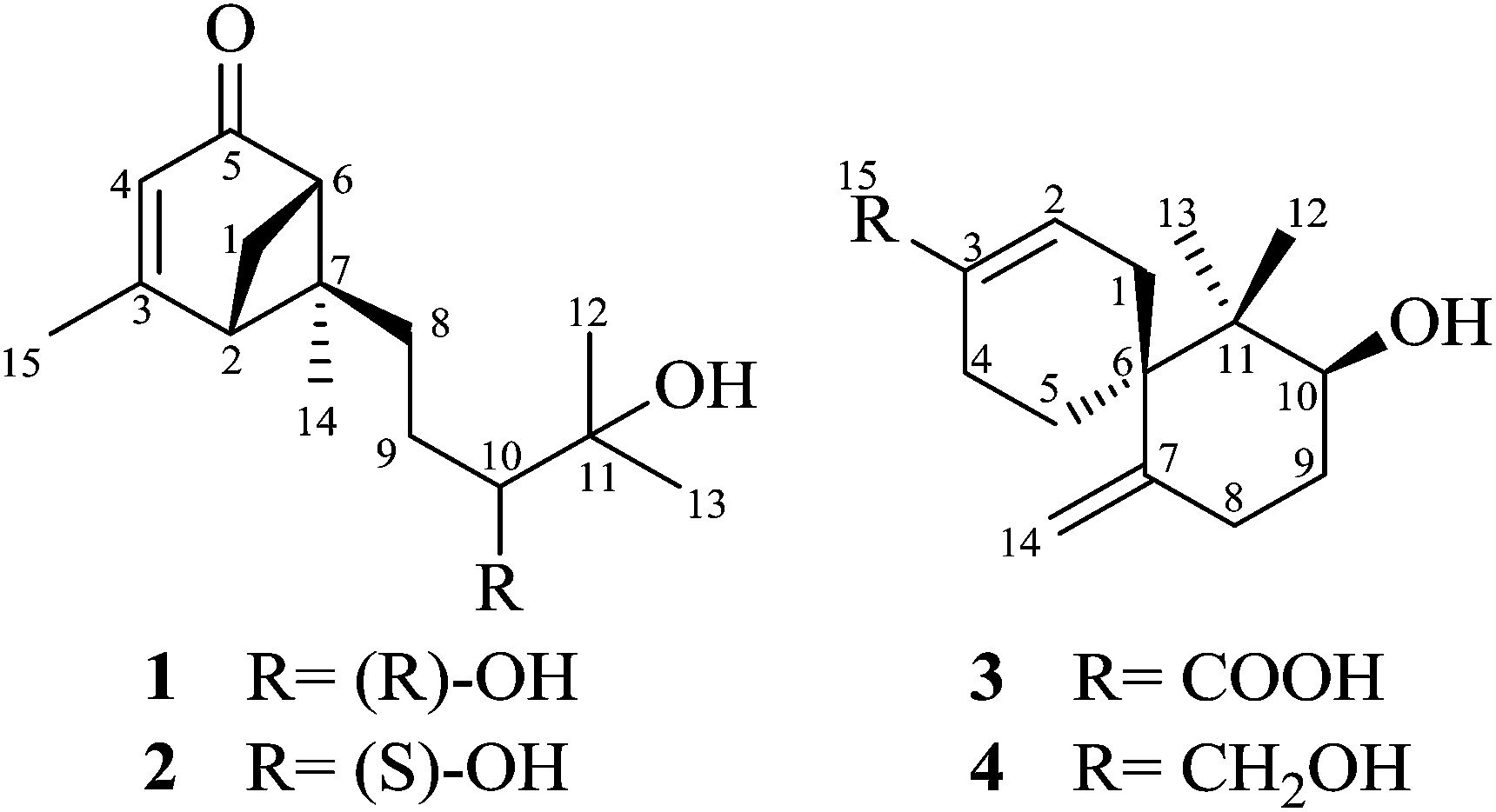
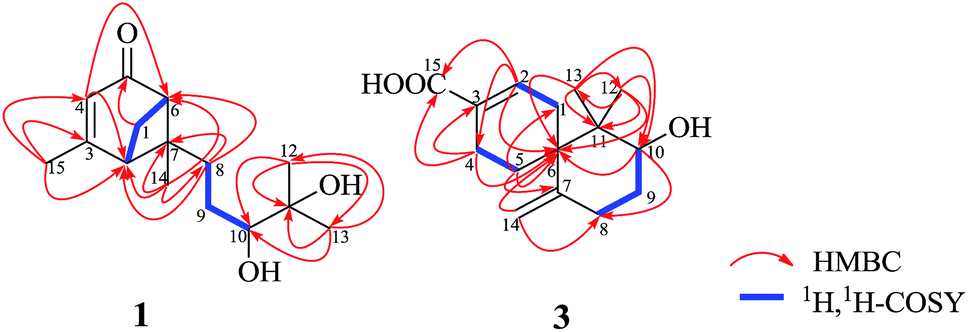
Xylariterpenoid A (1) was obtained as a oil with a molecular formula of C15H24O3 (four degrees of unsaturation), which was determined by HRESIMS at m/z 275.1615 [M + Na]+ (calcd for C15H24O3Na, 275.1623). The UV spectrum (252 nm) indicated the presence of an α,β-unsaturated carbonyl moiety. The IR spectrum showed bands corresponding to hydroxyl (3424 cm−1) and α,β-unsaturated carbonyl (1663 cm−1) group. The 1H NMR spectrum (Table 1) exhibited four methyls, three sp3 methylenes, three sp3 methines and one olefinic proton. The 13C NMR and DEPT spectra of 1 (Table 1) displayed 15 carbon signals consisting of four methyls, three methylenes, three sp3 methines, two sp3 quaternary carbons, two olefinic carbons, and one carbonyl carbon. The proton resonances were assigned to relevant carbon atoms through the HSQC experiment. Analysis of the 1H, 1H-COSY spectrum led to the identification of two isolated 1H spin-systems corresponding to the C-2–C-1–C-6 and C-8–C-9–C-10 subunits (Fig. 2). In addition, the HMBC correlations from H3-15 to C-2/C-3/C-4, from H-4 to C-2/C-6, from Ha-1 to C-5, from H3-14 to C-2/C-6/C-7/C-8, from H3-12 to C-10/C-11/C-13, from H3-13 to C-10/C-11/C-12, and from Ha-8 to C-2/C-6/C-7 deduced the planar structure of 1 with a bicyclo[3.1.1]heptane skeleton, which was shown in Fig. 2. Furthermore, the observed 4JH-2–H-6 (5.8 Hz) was consistent with the bridgehead-bridgehead coupling constant of the bicyclo[3.1.1]heptanes,4 which confirmed the above deduction.
Table 1 1H (400 MHz) and 13C (100 MHz) NMR spectroscopic data for xylariterpenoids A–D (1–4) in CDCl3 (δ in ppm, J in Hz)
| No. |
1 |
2 |
3 |
4 |
| δH |
δC |
δH |
δC |
δH |
δC |
δH |
δC |
| 1 |
2.07, d (9.1), a |
40.8 |
2.08, d (9.5), a |
40.8 |
2.17, m, a |
30.2 |
2.05, m, a |
28.9 |
| 2.80, dt (9.1, 5.3), b |
|
2.80, dt (9.5, 5.5), b |
|
2.31, m, b |
|
2.15, m, b |
|
| 2 |
2.45, br t (5.8) |
48.6 |
2.47, br t (5.7) |
48.4 |
7.09, m |
141.5 |
5.63, br s |
122.0 |
| 3 |
|
170.6 |
|
170.6 |
|
128.8 |
|
136.3 |
| 4 |
5.73, q (1.0) |
121.5 |
5.74, q (1.4) |
121.5 |
1.80, m, a |
21.4 |
1.67, m, a |
23.1 |
| |
|
|
|
2.31, m, b |
|
1.96, m, b |
|
| 5 |
|
204.4 |
|
204.2 |
1.48, m, a |
25.2 |
1.50, m, a |
25.4 |
| |
|
|
|
1.95, m, b |
|
1.87, m, b |
|
| 6 |
2.71, br t (5.8) |
55.5 |
2.71, br t (5.7) |
55.8 |
|
45.4 |
|
45.7 |
| 7 |
|
57.1 |
|
57.1 |
|
146.4 |
|
146.8 |
| 8 |
1.73, td (12.8, 4.0), a |
35.6 |
1.90, td (12.4, 4.5), a |
35.7 |
2.17, m, a |
30.0 |
2.17, m, a |
30.1 |
| 2.31, td (12.8, 4.0), b |
|
2.17, td (12.4, 4.5), b |
|
2.31, m, b |
|
2.33, m, b |
|
| 9 |
1.34, m, a |
26.5 |
1.34, m, a |
26.5 |
1.47, m, a |
31.9 |
1.45, m, a |
32.0 |
| 1.48, m, b |
|
1.50, m, b |
|
1.86, m, b |
|
1.82, m, b |
|
| 10 |
3.33, dd (10.3, 1.6) |
78.8 |
3.36, dd (10.4, 1.8) |
78.8 |
3.88, dd (11.9, 4.6) |
73.3 |
3.87, dd (11.9, 4.7) |
73.5 |
| 11 |
|
73.2 |
|
73.3 |
|
41.9 |
|
42.0 |
| 12 |
1.16, s |
23.3 |
1.17, s |
23.2 |
0.76, s |
15.1 |
0.73, s |
15.0 |
| 13 |
1.21, s |
26.5 |
1.23, s |
26.7 |
1.02, s |
20.4 |
1.01, s |
20.4 |
| 14 |
0.96, s |
19.0 |
0.98, s |
19.0 |
4.44, br s, a |
112.0 |
4.55, br s, a |
111.9 |
| |
|
|
|
4.92, br s, b |
|
4.92, br s, b |
|
| 15 |
2.00, d (1.0) |
23.6 |
2.01, d (1.4) |
23.6 |
|
171.7 |
3.92, br s |
67.2 |
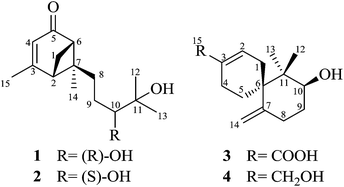 |
| | Fig. 1 Chemical structures of 1–4. | |
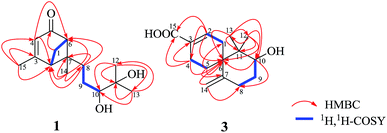 |
| | Fig. 2 Key HMBC and 1H, 1H–COSY of compounds 1 and 3. | |
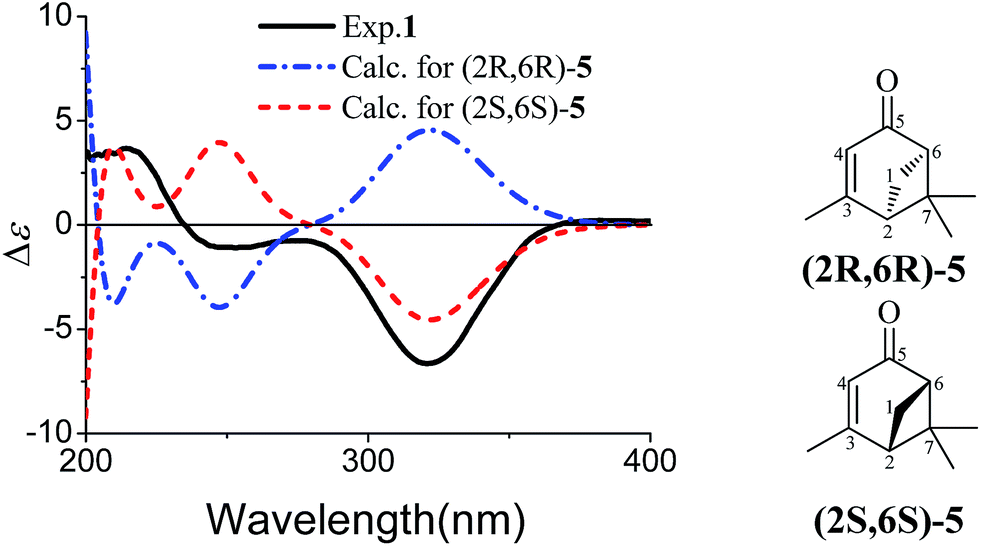
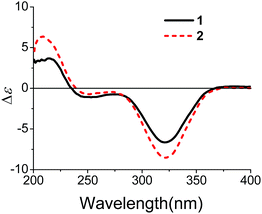
The partial relative configuration of 1 was established by the NOESY. The NOESY correlations between Hb-1 and Ha-8/Hb-8 indicated the relative configurations of C-2, C-6 and C-7 in 1 were 2S*, 6S*, and 7S*, respectively. The absolute configurations of C-2, C-6 and C-7 were determined by comparison of the experimental and the simulated circular dichroism (CD) spectra. Since the conformations of the flexible side chain in 1 had an insignificant effect on the CD spectrum, the simplified structures (2R,6R)-5 and (2S,6S)-5 were used for CD calculations (Fig. 3). The preliminary conformational analyses for a pair of enantiomers (2R,6R)-5 and (2S,6S)-5 were performed with Conflex version 7.0 via the MMFF94S force field. The corresponding minimum geometries were further fully optimized by DFT at the B3P86/6-31G(d) level as implemented in the Gaussian 09 program package. The obtained stable conformers were submitted to CD calculation by TDDFT [B3P86/6-311++G(2d,p)] method.5–8 The result showed that the experimental CD spectrum of 1 and the calculated CD spectrum of (2S,6S)-5 have similar negative Cotton effects (CEs) in the region of 320–350 nm (Fig. 3), which suggested that the absolute configurations of C-2, C-6 and C-7 in 1 were 2S, 6S, and 7S, respectively. In addition, the absolute configuration of the C-10 of 1 was determined by the in situ dimolybdenum CD method.9,10 The negative Cotton effect (∼310 nm), observed in the induced CD spectrum, permitted the assignment of 10R configuration according to the empirical rule proposed by Snatzke (Fig. 4). On the basis of the above deduction, the absolute configuration of 1 was assigned as 2S, 6S, 7S, 10R. Thus, the structure of 1 was established as (1S,5S,6S)-6-((R)-3,4-dihydroxy-4-methylpentyl)-4,6-dimethylbicyclo[3.1.1]hept-3-en-2-one, named as xylariterpenoid A.
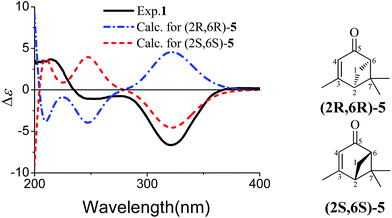 |
| | Fig. 3 Experimental CD spectrum of 1 in MeOH and calculated CD spectra of a pair of enantiomers of the corresponding simplified structures [(2R,6R)-5 and (2S,6S)-5]. | |
 |
| | Fig. 4 CD spectra of 1 (red) and 2 (blue) in DMSO containing Mo2(OAc)4 with the inherent CD spectrum subtracted. | |
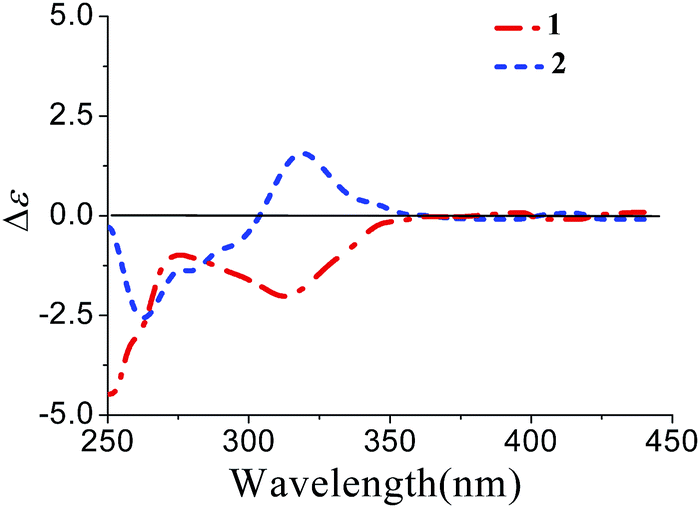
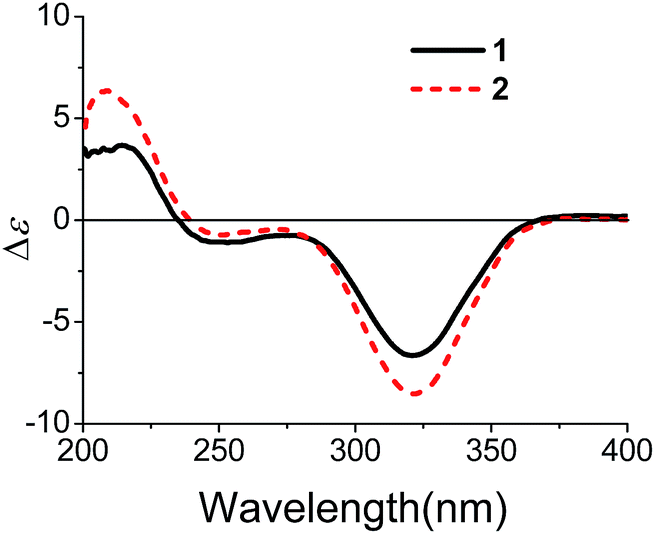
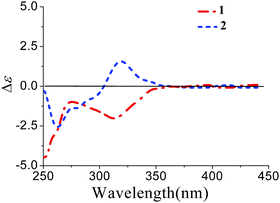
Xylariterpenoid B (2) was isolated as a oil with the same molecular formula as 1. Except for the chemical shifts of protons around C-10 (Ha-8, Hb-8, Hb-9, H-10, and H3-12, H3-13, H3-14), the 1H and 13C NMR data (Table 1) of 2 were similar to those of 1, which disclosed that both the skeleton and the functional groups presented in 2 were same as those of 1, and suggested that 2 was a epimer of 1 different at C-10. The 1H, 1H–COSY and HMBC correlations (the details are available in the ESI†) observed fully supported that 2 had the same planar structure as that of 1. The NOESY correlation between Hb-1 and Ha-8 indicated that the relative configurations of C-2, C-6 and C-7 were also the same as 1, which confirmed that 2 was a epimer of 1 different at C-10. In the CD spectrum, 2 showed the same Cotton effects as 1 (Fig. 5), indicating that the absolute configurations of C-2, C-6 and C-7 were S. However, the positive Cotton effect at around 310 nm, observed in the in situ dimolybdenum CD spectrum, permitted the assignment of 10S configuration for 2 (Fig. 4). On the basis of the above data, the absolute configuration of 2 was assigned as 2S, 6S, 7S, 10S. Thus, the structure of 2 was determined as (1S,5S,6S)-6-((S)-3,4-dihydroxy-4-methylpentyl)-4,6-dimethylbicyclo[3.1.1]hept-3-en-2-one, named as xylariterpenoid B.
 |
| | Fig. 5 CD spectra of 1 and 2 in MeOH. | |
Xylariterpenoid C (3) was assigned as C15H22O3 (five degrees of unsaturation) on the basis of its HRESIMS at m/z 273.1465 [M + Na]+ (calcd for C15H22O3Na, 273.1467). The IR spectrum showed bands corresponding to hydroxyl (3439 cm−1). The 1H NMR spectrum (Table 1) exhibited two methyls, five sp3 methylenes, one oxygenated sp3 methine and three olefinic protons. The 13C NMR and DEPT spectra of 3 (Table 1) displayed 15 carbon signals consisting of two methyls, five sp3 methylenes, one oxygenated sp3 methine, two sp3 quaternary carbons, four olefinic carbons, and one carbonyl carbon. The proton resonances were assigned to relevant carbon atoms through the HSQC experiment. Interpretation of the 1H, 1H-COSY spectrum of 3 identified three isolated proton spin systems, which were C-1–C-2, C-4–C-5, and C-8–C-9–C-10 (Fig. 2). Combined with the analysis of 1H, 1H-COSY spectrum, the HMBC correlations from H3-13 to C-6/C-10/C-11/C-12, from H3-12 to C-6/C-10/C-11/C-13, from H2-14 to C-6/C-8, from H-2 to C-4/C-6/C-15, from Hb-4 to C-3/C-6/C-15, from Hb-5 to C-1 and from Ha-5 to C-6/C-7 completed the planar structure of 3 (Fig. 1).
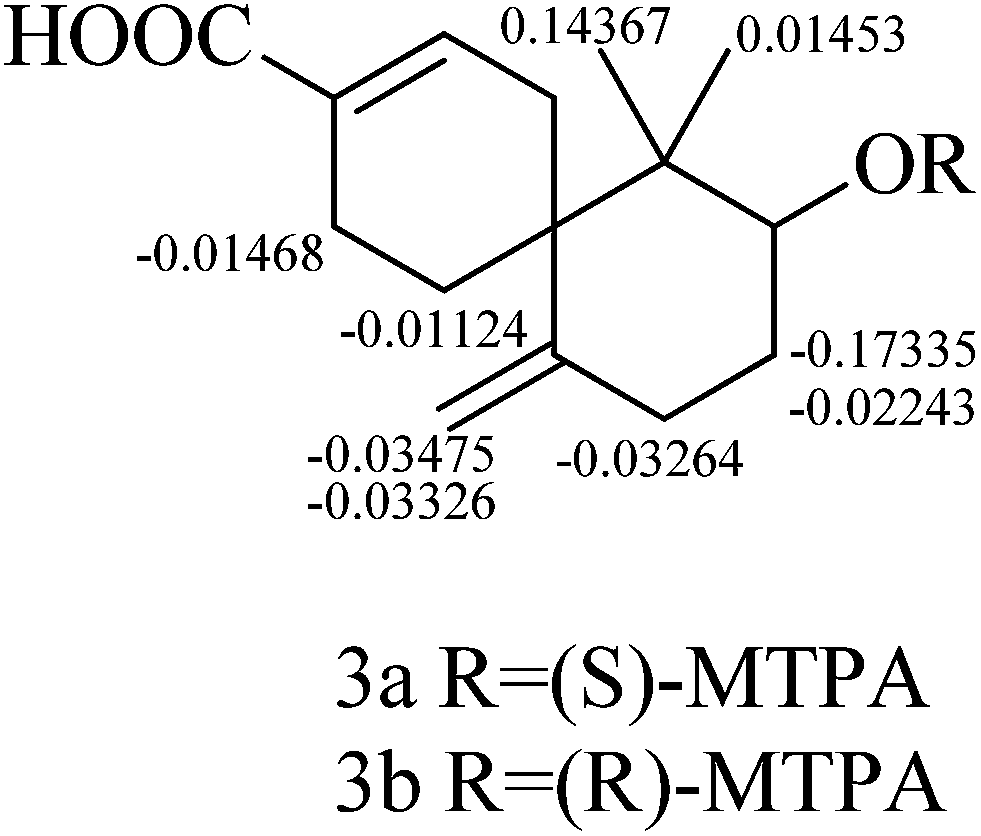
The relative configuration of 3 was established by the NOESY data analysis. The orientation of H-10 was the same as those of H2-5 and H3-13, which was determined by the NOESY correlations between H-10 and H3-13/Hb-5. Thus, the relative configuration of 3 was designated as 6R*, 10S*. The absolute configuration of 3 was determined by the modified Mosher method.11 The Δδ values of the (S)- and (R)-MTPA esters (3a and 3b) indicated the S configuration for C-10 (Fig. 6). On the basis of the above deduction, the absolute configuration 6R, 10S was proposed for 3. Therefore, the structure of 3 was elucidated as (6R,8S)-8-hydroxy-7,7-dimethyl-11-methylenespiro[5.5]undec-2-ene-3-carboxylic acid, named as xylariterpenoid C.
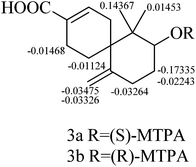 |
| | Fig. 6 Δδ(δS − δR) values (in ppm) obtained for (S)- and (R)-MTPA esters (3a and 3b). | |
Xylariterpenoid D (4) was isolated as a crystal. Its HRESIMS analysis gave a quasi-molecular ion peak at m/z 259.1667 [M + Na]+ (calcd for C15H24O2Na, 259.1674), 14 mass units less than 3. The 1D NMR data of 4 revealed its structural features were similar to those of 3, except that the resonance for C-15 [δC 171.7 (C-15)] was replaced by an oxygenated methylene group [δC/H 67.2 (C-15)/3.92 (br s, H2-15)]. At the same time, the 1H, 1H-COSY and HMBC correlations (the details are available in the ESI†) observed fully supported the locations of functional groups. Thus, the planar structure of 4 was established as shown in Fig. 1.
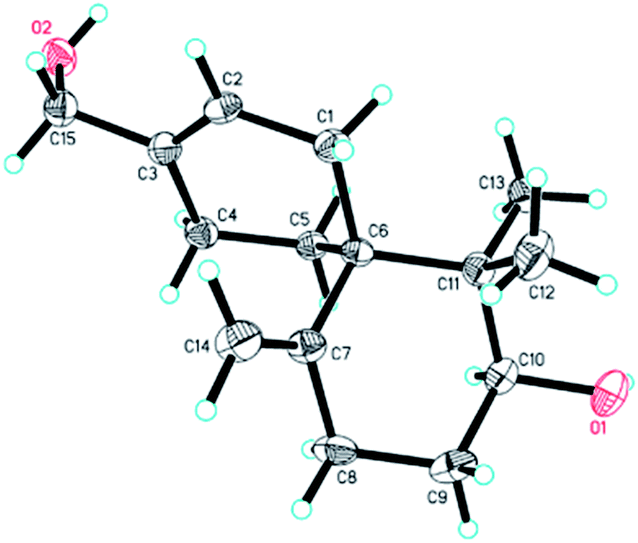
The relative configuration of 4 was established on the basis of the NOESY data. The NOESY correlation between H-10 and Hb-5 indicated that 4 had the same relative configuration with 3. In the meanwhile, the relative configuration of 4 was confirmed by single-crystal X-ray crystallographic analysis (Fig. 7). Furthermore, the values of the Flack parameter (−0.1(3)) and the Hooft y parameter 0.04(16) allowed the assignment of the absolute configuration of 4 as 6R, 10S. Therefore, the structure of 4 was determined as (2S,6R)-9-(hydroxymethyl)-1,1-dimethyl-5-methylenespiro[5.5]undec-8-en-2-ol, named as xylariterpenoid D.§
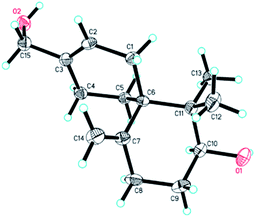 |
| | Fig. 7 X-ray crystal structure of 4. | |
Biological activities
All compounds were tested for the cytotoxicities against HL-60, A-549, MCF-7, SMMC-7721, and SW-480 human cancer cell lines using MTT method. However, compounds 1–4 exhibited no significant cytotoxicity, with IC50 > 40 μM.
Discussion and conclusion
Sesquiterpenes incorporating the bergamotane skeleton have been reported from fungi (such as Ampulliferina sp.,12 Ampulliferina-like sp. no. 27,13,14 Aspergillus fumigatus,15,16 Massarina tunicate,17Penicillum expansum,18 and Podospora decipiens19) and plants (such as Tanacetum vulgare,20 and Lepidolaena clavigera21,22). Compounds 1 and 2 are the first examples of the bergamotane-type sesquiterpenes with carbonyl at C-5 position. In addition, the previously reported chamigrane-type sesquiterpenes are halogenated analogues, which were isolated from red algae.22 Compounds 3 and 4 are new members of the non-halogenated chamigrane-type sesquiterpenes. Although all new compounds exhibited no significant cytotoxicity, further studies still need to elucidate their promising bioactivity.
Acknowledgements
This work was financially supported by grants from the Ministry of Science and Technology of China (2012ZX09301002-003001006), the National Natural Science Foundation of China (81422054, 81373306, 81202441), the Guangdong Natural Science Funds for Distinguished Young Scholar (S2013050014287), Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (Hao Gao, 2014), and the high-performance computing platform of Jinan University.
Notes and references
- M. Stadler and V. Hellwig, Recent Res. Dev. Phytochem., 2005, 9, 41–93 CAS.
- A. J. S. Whalley and R. L. Edwards, Can. J. Bot., 2007, 73, 802–810 CrossRef PubMed.
-
(a) G. D. Chen, Y. J. Li, H. Gao, Y. Chen, X. X. Li, J. Li, L. D. Guo, Y. Z. Cen and X. S. Yao, Planta Med., 2012, 78, 1683–1689 CrossRef CAS PubMed;
(b) J. W. He, G. D. Chen, H. Gao, F. Yang, X. X. Li, T. Peng, L. D. Guo and X. S. Yao, Fitoterapia, 2012, 83, 1087–1091 CrossRef CAS PubMed;
(c) F. Yang, G. D. Chen, H. Gao, X. X. Li, Y. Wu, L. D. Guo and X. S. Yao, J. Asian Nat. Prod. Res., 2012, 14, 1059–1063 CrossRef CAS PubMed;
(d) G. D. Chen, Y. Chen, H. Gao, L. Q. Shen, Y. Wu, X. X. Li, Y. Li, L. D. Guo, Y. Z. Cen and X. S. Yao, J. Nat. Prod., 2013, 76, 702–709 CrossRef CAS PubMed;
(e) Q. C. Zheng, G. D. Chen, M. Z. Kong, J. Y. Cui, X. X. Li, Z. Y. Wu, L. D. Guo, Y. Z. Cen, Y. Z. Zheng and H. Gao, Steroids, 2013, 78, 896–901 CrossRef CAS PubMed;
(f) F. Ye, G. D. Chen, J. W. He, X. X. Li, X. Sun, L. D. Guo, Y. Li and H. Gao, Tetrahedron Lett., 2013, 54, 4551–4554 CrossRef CAS PubMed;
(g) G. D. Chen, Y. R. Bao, Y. F. Huang, D. Hu, X. X. Li, L. D. Guo, J. Li, X. S. Yao and H. Gao, Fitoterapia, 2014, 92, 252–259 CrossRef CAS PubMed;
(h) Q. C. Zheng, M. Z. Kong, Q. Zhao, G. D. Chen, H. Y. Tian, X. X. Li, L. D. Guo, Y. Z. Zheng and H. Gao, Fitoterapia, 2014, 93, 126–131 CrossRef CAS PubMed;
(i) H. Xiong, G. K. Xiao, G. D. Chen, H. R. Chen, D. Hu, X. X. Li, S. W. Zhong, L. D. Guo, X. S. Yao and H. Gao, RSC Adv., 2014, 4, 24295–24299 RSC;
(j) H. Zhao, G. Q. Wang, X. P. Tong, G. D. Chen, Y. F. Huang, J. Y. Cui, M. Z. Kong, L. D. Guo, Y. Z. Zheng, X. S. Yao and H. Gao, Fitoterapia, 2014, 98, 77–83 CrossRef CAS PubMed.
- R. B. Bates and V. P. Thalacker, J. Org. Chem., 1968, 33, 1730–1732 CrossRef CAS.
- H. J. Zhu, Modern Organic Stereochemistry, Science Presses, Beijing, 2009 Search PubMed.
- J. Ren, J. X. Jiang, L. B. Li, T. G. Liao, R. R. Tian, X. L. Chen, S. P. Jiang, C. J. Pittman and H. J. Zhu, Eur. J. Org. Chem., 2009, 3987–3991 CrossRef CAS.
- X. N. Li, Y. Zhang, X. H. Cai, T. Feng, Y. P. Liu, Y. Li, J. Ren, H. J. Zhu and X. D. Luo, Org. Lett., 2011, 13, 5896–5899 CrossRef CAS PubMed.
- S. D. Zhao, L. Shen, D. Q. Luo and H. J. Zhu, Curr. Org. Chem., 2011, 15, 1843–1862 CrossRef CAS.
- L. D. Bari, G. Pescitelli, C. Pratelli, D. Pini and P. Salvadori, J. Org. Chem., 2011, 66, 4819–4825 CrossRef PubMed.
- M. Gorecki, E. Jablonska, A. Kruszewska, A. Suszczynska, Z. Urbanczyk-Lipkowska, M. Gerards, J. W. Morzycki, W. J. Szczepek and J. Frelek, J. Org. Chem., 2007, 72, 2906–2916 CrossRef CAS PubMed.
- I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092–4096 CrossRef CAS.
- Y. Kimura, H. Nakajima, T. Hamasaki, T. Matsumoto, Y. Matsuda and A. Tsuneda, Agric. Biol. Chem., 1990, 54, 813–814 CrossRef CAS.
- Y. Kimura, T. Matsumoto, H. Nakajima, T. Hamasaki and Y. Matsuda, Biosci., Biotechnol., Biochem., 1993, 57, 687–688 CrossRef CAS.
- S. Nozoe, H. Kobayashi and N. Morisaki, Tetrahedron Lett., 1976, 17, 4625–4626 CrossRef.
- F. A. Macias, R. M. Varela, A. M. Simonet, H. G. Cutler, S. J. Cutler and R. A. Hill, Tetrahedron Lett., 2003, 44, 941–943 CrossRef CAS.
- H. Oh, J. B. Gloer and C. A. Shearer, J. Nat. Prod., 1999, 62, 487–501 CrossRef PubMed.
- M. Massias, S. Rebuffat, L. Molho, A. Chiaroni, C. Riche and B. Bodo, J. Am. Chem. Soc., 1990, 112, 8112–8115 CrossRef CAS.
- Y. Che, J. B. Gloer, B. Koster and D. Malloch, J. Nat. Prod., 2002, 65, 916–919 CrossRef CAS PubMed.
- A. Chandra, L. N. Misra and R. S. Thakur, Phytochemistry, 1987, 26, 3077–3078 CrossRef CAS.
- N. B. Perry, E. J. Burgess, L. M. Foster and P. J. Gerard, Tetrahedron Lett., 2003, 44, 1651–1653 CrossRef CAS.
- N. B. Perry, E. J. Burgess, L. M. Foster, P. J. Gerard, M. Toyota and Y. Asakawa, J. Nat. Prod., 2008, 71, 258–261 CrossRef CAS PubMed.
- N. F. Sergei, K. S. Larisa, I. K. Anatoly, G. L. Ekaterina and A. S. Valentin, Tetrahedron Lett., 2000, 41, 1979–1982 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: The general experimental procedure, fungus material, extraction and isolation, spectroscopic data of 1–4, quantum chemical CD calculation of 5, single-crystal X-ray data of 4, the in situ dimolybdenum CD method, preparation of (S)- and (R)-MTPA esters of 3 (3a and 3b), cytotoxicity assay, and 1D and 2D NMR spectra of compounds 1–4. CCDC 1023174. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra10365c |
| ‡ These authors have contributed equally to this work. |
§ Crystal data of xylariterpenoid D (4): data were collected using a Sapphire CCD with a graphite monochromated Cu Kα radiation, λ = 1.54184 Å at 173.00 (10) K. Crystal data: C15H24O2, M = 236.34, orthorhombic, space group P212121; unit cell dimensions were determined to be a = 6.5259(3) Å, b = 13.1402(5)Å, c = 15.6863(7) Å, α = 90.00°, β = 90.00°, γ = 90.00°, V = 1345.13(10) Å3, Z = 4, Dx = 1.167 g cm−3, F (000) = 520, μ (Cu Kα) = 0.586 mm−1. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 952 reflections were collected until θmax = 62.93°, in which independent unique 1950 reflections were observed [F2 > 4σ(F2)]. The final refinement gave R = 0.0363, RW = 0.0909, S = 1.079, Flack = −0.1(3), and Hooft y = 0.04(16). 952 reflections were collected until θmax = 62.93°, in which independent unique 1950 reflections were observed [F2 > 4σ(F2)]. The final refinement gave R = 0.0363, RW = 0.0909, S = 1.079, Flack = −0.1(3), and Hooft y = 0.04(16). |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.