
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
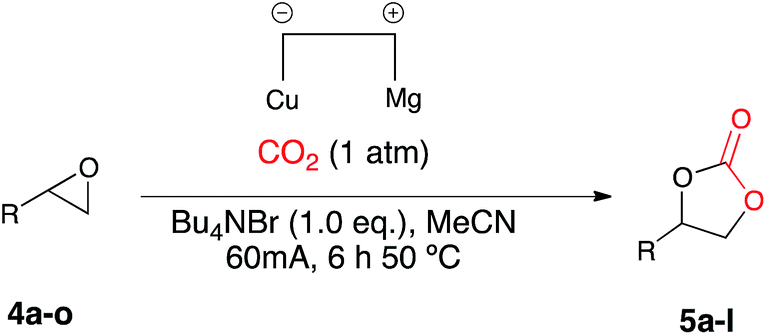
‘Ring-expansion addition’ of epoxides using applied potential: an investigation of catalysts for atmospheric pressure carbon dioxide utilization
Benjamin R.
Buckley
*,
Anish P.
Patel
and
K. G. Upul
Wijayantha
*
Department of Chemistry, Loughborough University, Loughborough, Leicestershire, LE11 3TU, UK. E-mail: b.r.buckley@lboro.ac.uk
First published on 29th October 2014
Abstract
Interest in the use of CO2 as a feedstock for preparation of value-added compounds has dramatically increased over the past decade. We herein investigate the insertion of atmospheric pressure carbon dioxide under electrosynthetic conditions into epoxides. The initial investigations involved the use of either a nickel or copper based catalyst, however, upon further optimisation and careful selection of electrode materials we found that these catalysts were not required for efficient carbon dioxide insertion to occur.
Introduction
Anticipated pressures on fossil fuel resources in the coming decades mean that raw materials for the chemical industry must be found that can reduce our reliance on fossil fuel-based feedstocks.1 Hence, the development of new production processes for chemical syntheses from renewable resources is a challenge that should be a priority for all major economies.2 Research into/application of carbon dioxide utilization (CDU) technologies has recently gained increased interest.3 CDU offers an attractive alternative to the well-established carbon dioxide storage (CCS) processes, however, scale of production means that CDU is unlikely to significantly reduce anthropogenic carbon dioxide emissions, but waste carbon dioxide under CDU protocols can be converted into useful chemicals or fuels.Carbon dioxide is abundant, cheap and non-toxic when compared to other C1 building blocks, such as phosgene, and has been used in the manufacture of salicylic acid, urea and cyclic carbonates for 50–100 years. However, due to carbon dioxides relative inertness these processes are significantly energy demanding with reactions taking place at high temperatures and pressures. Therefore if CDU technologies are to become viable processes with reliance on very little energy input are required, hence processes at mild temperatures (<100 °C) and atmospheric pressures should be targeted.
The chemical reduction of CO2 has received considerable interest in the last few years, both as a method for recycling CO2 in industrial waste streams and as well as a route to useful chemical products. For example, cyclic carbonates are widely used in the manufacture of products including solvents, paint-strippers, biodegradable packaging, as well as having applications in the chemical industry. Cyclic carbonates also have potential for use in the manufacture of a new class of efficient anti-knocking agents in petrol.4
The insertion of carbon dioxide into epoxides to afford either 5-membered ring cyclic carbonates5,6 or the corresponding polycarbonate7 has attracted considerable attention due to the industrial application of the products arising from these reactions. For example, North and co-workers have employed bimetallic aluminium salen complexes incorporating a ammonium bromide co-catalyst such as 1 (Scheme 1) to prepare cyclic carbonates in good yield from not only highly purified carbon dioxide but also directly from a fossil fuel power station stack by use of a supported salen catalyst and flow reactor.8 These reactions take place at mild temperatures and atmospheric pressures.
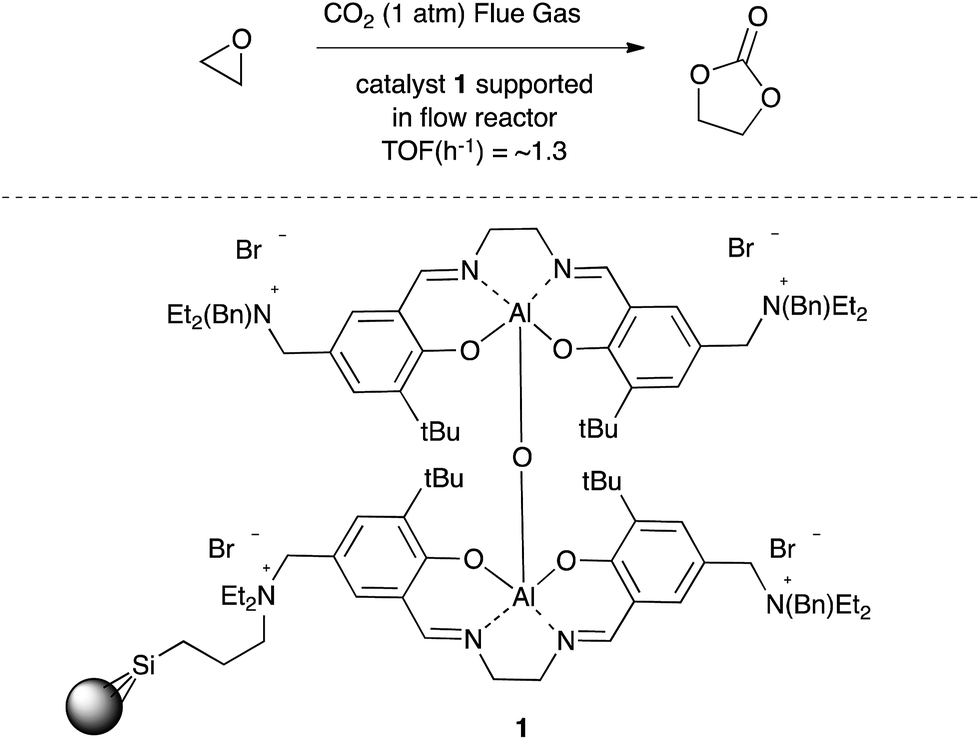 | ||
| Scheme 1 One of North's catalysts for cyclic carbonate synthesis using flue gas. | ||
Other catalytic systems have also been successfully employed for example, Kleij and co-workers have recently reported the synthesis of 5-membered cyclic carbonates in excellent yield using either an iron, aluminium or vanadium based catalyst at 10−2 atm CO2.9
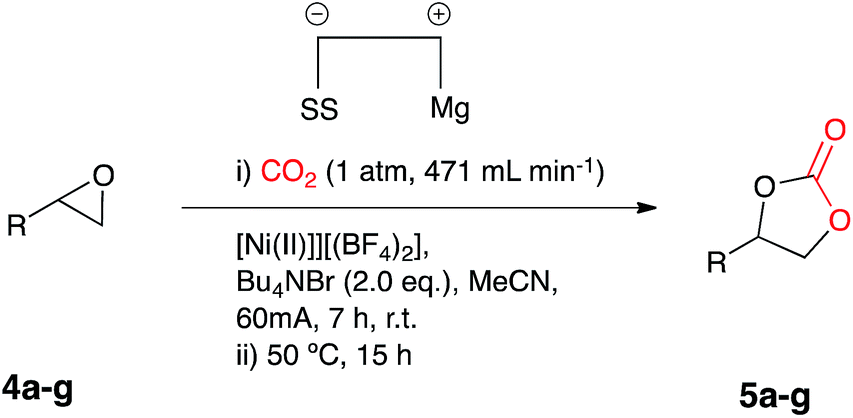
Duñach reported in the late 1990's the nickel-catalysed electrochemical carboxylation of epoxides using a nickel(II) cyclam complex (10 mol%) in DMF with a magnesium anode and KBr as electrolyte.10 This was an attractive system for fixing CO2, when one considers that typically the reaction of epoxides with CO2 for the preparation of cyclic carbonates is generally carried out at high temperatures (>100 °C) and elevated CO2 pressures (>40 atm) in the presence of a transition metal as catalyst.
Results and discussion
We were interested in developing a low energy alternative in line with our current research portfolio aimed at electrosynthesis11 coupled with semiconductor photoelectrodes to drive light assisted electrosynthetic reactions.12 We were therefore attracted to the possibility of employing an electrosynthetic system as this type of process could be designed to be cost neutral in terms of energy consumption if combined with a suitable solar powered energy source and unlike many metal catalysed processes be able to run at ambient temperature and atmospheric pressure carbon dioxide.Initial investigations of a nickel(I) catalysed system
Our initial aims were to improve the Duñach system by running the reactions with cheaper and more accessible catalysts and in a more user-friendly reaction medium. Based on conditions reported in literature the catalysts required for electrochemical carboxylation: [Ni(bipy)3][(BF4)2] 2 and Ni[(phen)3][(BF4)2] 3 were synthesised.13 Reactions were screened using styrene oxide 4a as substrate and run in acetonitrile in a single compartment cell containing a magnesium anode, a stainless steel cathode and Bu4NBr supporting electrolyte (2 eq.), at constant current: 60 mA (7 h), with constant CO2 flow (471 mL min−1), in the presence of the nickel catalyst (10 mol%) followed by heating at 50 °C (12 h, Scheme 2). The reaction proceeded in the presence of the nickel catalysts 2 and 3, forming the target cyclic carbonate 5a, in moderate to good conversion (65 and 53% respectively). Attempts to increase the conversion using catalyst 2 were successful when the heating time was increased to 18 h affording the carbonate in 81% conversion by 1H NMR spectroscopy (replacement of Bu4NBr with Bu4NI resulted in only 67% conversion).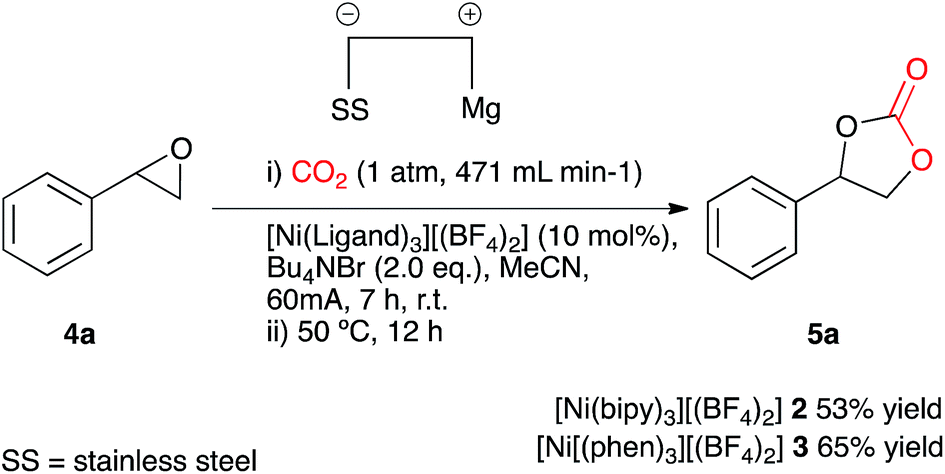 | ||
| Scheme 2 Initial screening conditions. | ||
In order to improve the system and move towards cheaper more accessible catalysts, [Ni(CH3CN)4][(BF4)2] 6 was employed in the electrocarboxylation of styrene oxide 4a. A test reaction (Scheme 3) involved electrolysis in acetonitrile in a single compartment cell containing a magnesium anode, a stainless steel cathode and Bu4NBr supporting electrolyte (2 eq.), at constant current: 60 mA (7 h), with constant CO2 flow (471 mL min−1), in the presence of the nickel catalyst 6 (10 mol%) followed by heating at 50 °C (12 h), forming cyclic carbonate 5a in moderate conversion (50%). A distinct colour change of the reaction solution on addition of the supporting electrolyte from blue to deep blue was observed during this reaction and it was thus believed that the active catalyst was the tetrahedral complex [Ni(Br)4], formed from a ligand exchange reaction between bromine ions of the supporting electrolyte and the co-ordinated acetonitrile ligands. Furthermore stability issues of reported cross bromo/acetonitrile complexes,14 suggest that bromine ions have a stronger affinity towards Ni(II) in comparison to acetonitrile ligands, eliminating the possibility of a cross complex. Resulting reactions involved direct addition of the nickel reagent, [Ni(II)][(BF4)2] 7 to the reaction solvent containing additional supporting electrolyte, Bu4NBr (Ni2+ + 4Br− → Ni(Br)48) to allow in situ active catalyst formation.
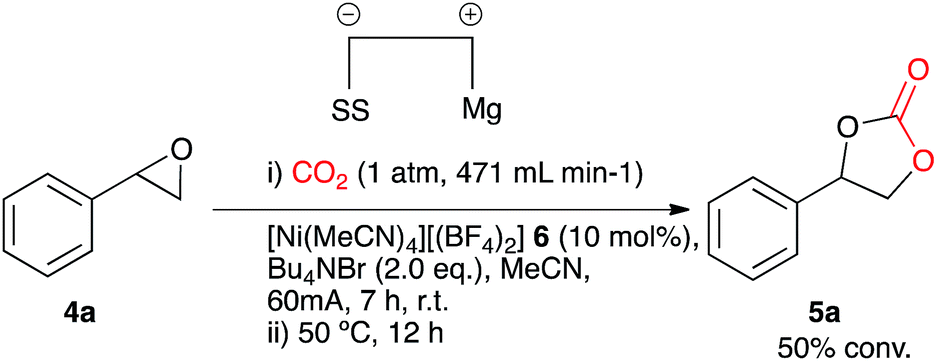 | ||
| Scheme 3 Nickel catalysed conditions. | ||
The optimum catalyst loading was also investigated, this time using 1,2-epoxyoctadecane 4b as a test substrate. Electrocarboxylation under the test reaction conditions in the presence of nickel reagent 7 at 10, 15, 20 and 50 mol% loading, Bu4NBr (2.4, 2.6, 2.8 and 4 equivalents) and heating for 15 hours, resulted in the corresponding cyclic carbonate 5b, with conversion increasing with catalyst loading. Although 50 mol%, gave the highest conversion (90%) in comparison to 10 mol% (55% conversion).
In order to further increase the conversion of starting material, the flow rate of CO2 entering the reaction system was studied. Electrocarboxylation of 4a under the test reaction conditions in the presence of CO2 at either 89, 287, or 471 mL min−1, nickel reagent 7 (50 mol%), Bu4NBr (4 eq.) and heating for 15 hours, produced the cyclic carbonate 5a, with conversion generally increasing with flow rate. A flow rate of 471 mL min−1 resulted in the highest conversion (88%).
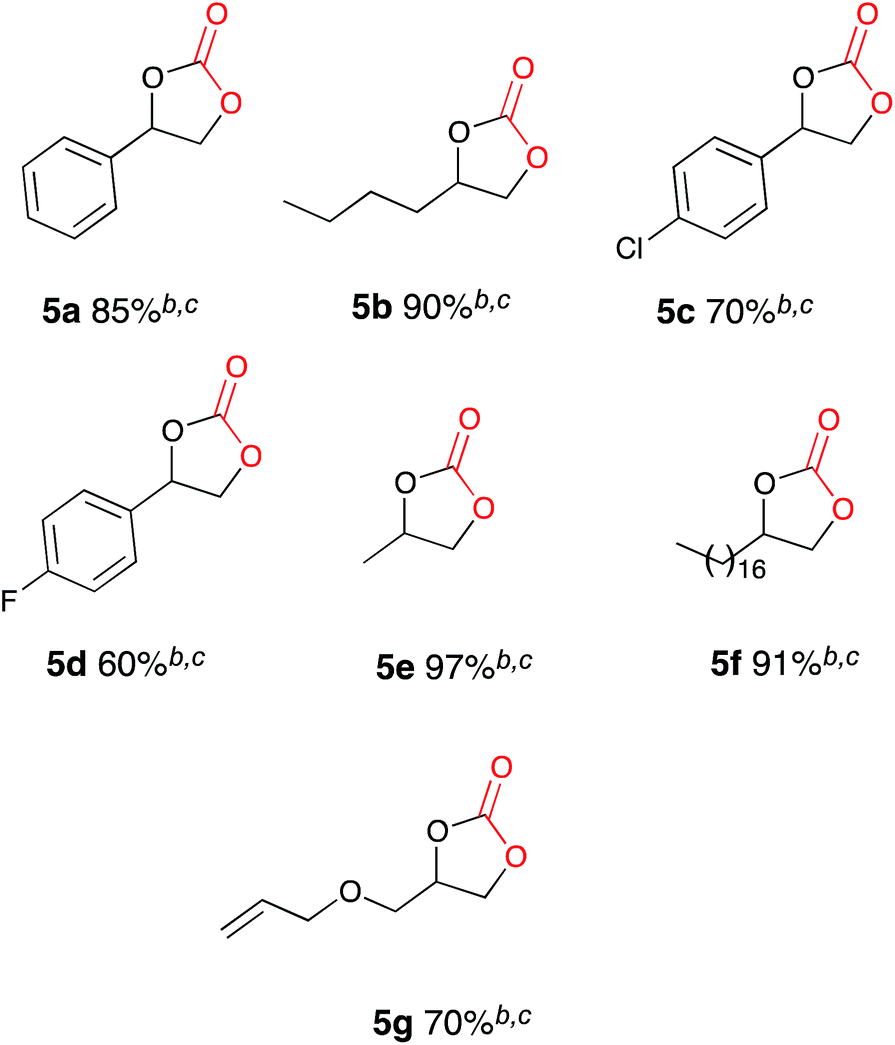
Once the optimum reaction conditions were established: 50 mol% loading of nickel reagent 7, 15 h heating and 471 mL min−1 CO2 flow rate, and in conjunction with existing conditions: open single compartment cell, magnesium anode, stainless steel cathode, acetonitrile solvent, Bu4NBr (4 eq.) and 7 h constant current electrolysis at 60 mA, a range of epoxide substrates were employed in order to determine the scope of the reaction conditions (Table 1). Electrocarboxylation of epoxides 4a–4g led to the formation of the target cyclic carbonates 5a–5g in moderate to excellent conversion (53–93%). Substrates 4c and 4d gave moderate conversion attributed to the electron withdrawing properties of the aromatic ring substituents, although both Cl− and F− substituents are mesomerically donating (+M) they are inductively withdrawing (−I), thus possibly decreasing the reactivity of both substrates to nucleophiles or Lewis acid interactions.
|
|
|---|
| a General reaction conditions: [Ni(II)][(BF4)2] (1.0 mmol), epoxide (2.0 mmol), stainless steel cathode, magnesium anode, Bu4NBr (8 mmol), CO2, 60 mA, 25 °C, 7 h, followed by: 50 °C, 15 h. b Conversion evaluated from the 1H NMR spectrum by integration of the epoxide vs. cyclic carbonate signals. c The cyclic carbonate was the only observed product by 1H NMR spectroscopy. |
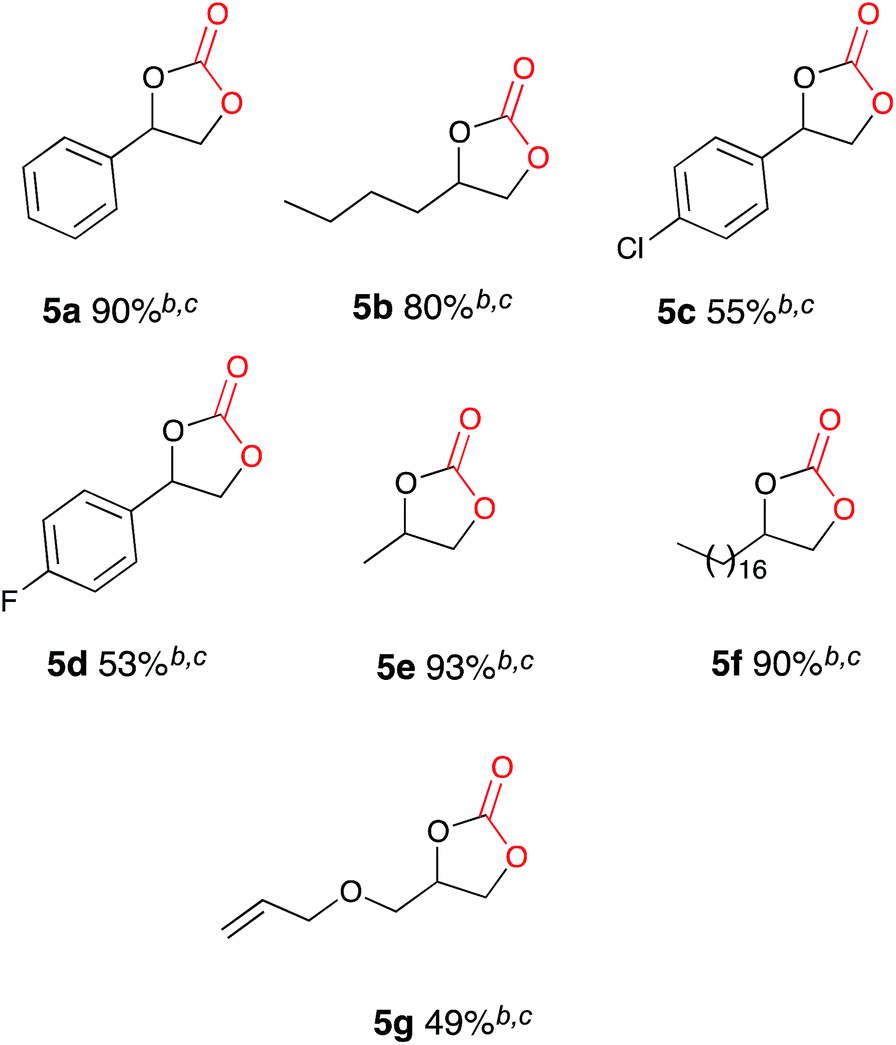
|
In order to prove the requirement of CO2, constant CO2 flow, Bu4NBr addition (4 eq.) and in situ formation of nickel catalyst 7, control experiments were carried out, involving the attempted synthesis of cyclic carbonate 5b from test substrate 4b under optimum conditions. Electrocarboxylation with a CO2 flush (1 h) prior to electrolysis and the absence of constant CO2 flow during the reaction, resulted in minimal conversion (5%), whereas in the absence of CO2 altogether no product formation could be detected by 1H NMR spectroscopy. Electrocarboxylation using lower loadings of Bu4NBr (1 eq.) resulted in decreased conversion (25%) in comparison to using 4 equivalents (90%), and finally electrocarboxylation using nickel catalyst 8 synthesised in CH3CN or EtOH resulted in comparable conversion (85% and 86%) to the in situ formed complex (90%).
Investigations of a copper(I) based system
In search of more effective catalysts for the synthesis of cyclic carbonates led to the identification of [Cu(CH3CN)4][BF4] 9. This coordination complex is cheaper and less toxic in comparison to the analogous nickel species. Electrocarboxylation of the test substrate 4a in acetonitrile in a single compartment cell containing a magnesium anode, stainless steel cathode and Bu4NBr supporting electrolyte (2.4 eq.), at constant current: 60 mA (7 h) with constant CO2 flow (471 mL min−1) in the presence of copper catalyst 9 (10 mol%) followed by heating (12 h) resulted in cyclic carbonate 5a with good conversion (75%, Scheme 4). It appeared, however, that the above mentioned catalyst was not the active catalyst due to a distinct colour change of the reaction solution on addition of the supporting electrolyte from colourless to purple. Based on previously reported systems it was thought that the active catalyst could be the tetrahedral complex [Cu(Br)4] 10.15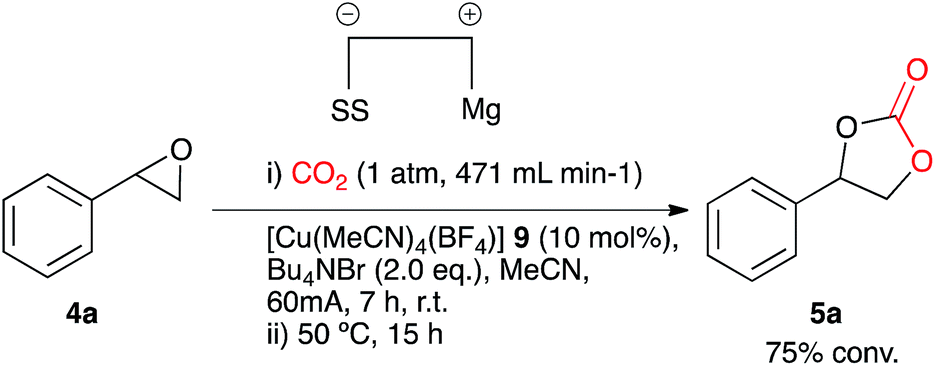 | ||
| Scheme 4 Copper(I) catalysed electrocarboxylation of styrene oxide. | ||
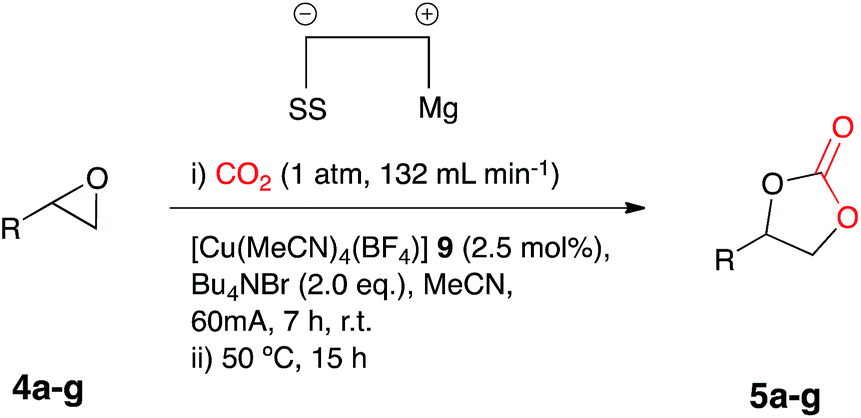
The reactions employing both nickel and copper based catalysts/reagents employed the use of an open reaction vessel during the electrocarboxylation process, thus an extremely high CO2 flow rate was required in order to achieve a higher conversion of the epoxide starting material to the cyclic carbonate product. In an effort to reduce the CO2 flow rate, the experimental set-up was altered to a pseudo-sealed reaction system, where CO2 had a single inlet and outlet point, allowing the measurement and potential recovery of CO2 leaving the system. Electrocarboxylation of 4a using the conditions described above, using a pseudo-sealed reaction vessel and a decreased CO2 flow rate of 132 mL min−1, formed the corresponding cyclic carbonate 5a in a similar conversion (76%) to the open reaction system (75%, flow rate: 471 mL min−1).
The optimum catalyst loading for this process was investigated and electrocarboxylation of 4a under the initially employed conditions in a pseudo sealed reaction vessel, in the presence of copper catalyst 9 at 2.5, 5 and 10 mol%, Bu4NBr (2.1, 2.2 and 2.4 eq.) and a CO2 flow rate of 132 mL min−1, resulted in the formation of the cyclic carbonate 5a, with conversion decreasing with catalyst loading. Unusually the lowest catalyst loading of 2.5 mol% resulted in a slightly higher conversion (85%), in comparison to 5 mol% (81%) and 10 mol% (78%).
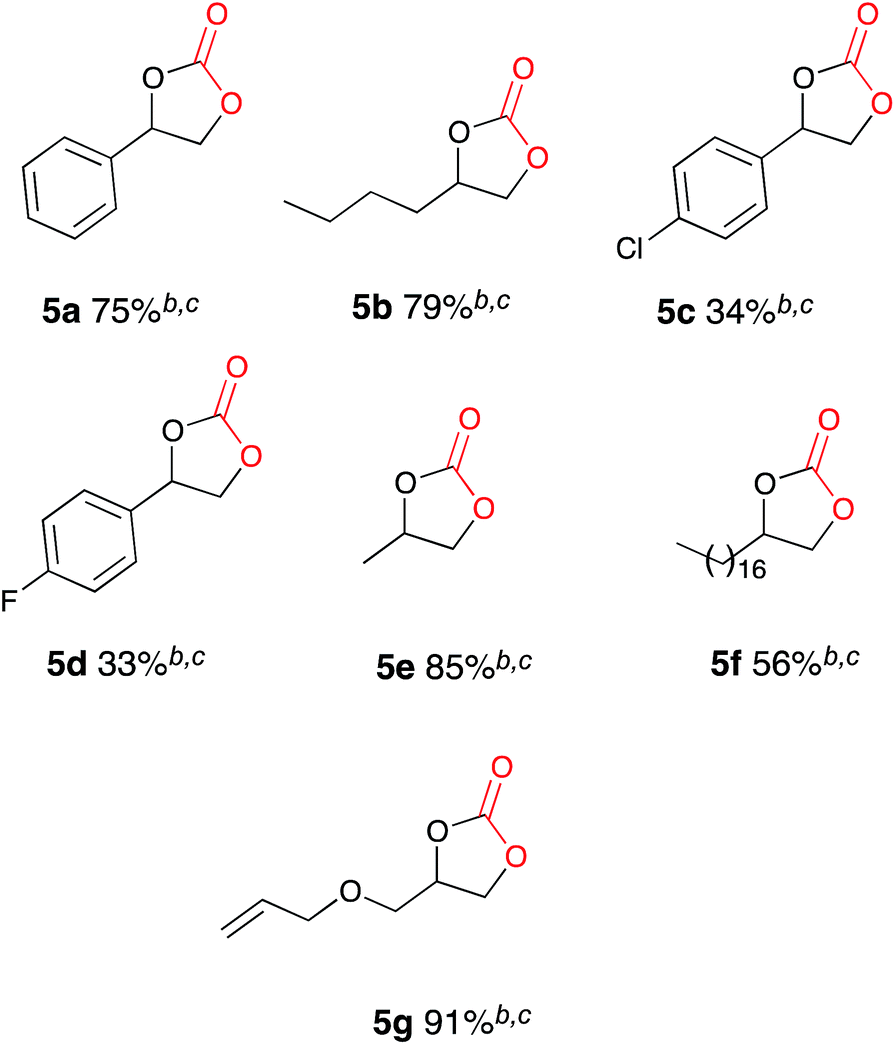
Once the optimum reaction conditions were established: pseudo-sealed single compartment cell, 2.5 mol% loading of copper catalyst 9, 132 mL min−1 CO2 flow rate, and in conjunction with the existing conditions: magnesium anode, stainless steel cathode, acetonitrile solvent, Bu4NBr (2.1 eq.), 7 h constant current electrolysis at 60 mA and heating for 12 h, a range of epoxide substrates were employed in order to determine the scope of the reaction conditions (Table 2). Electrocarboxylation of epoxides 4a–4g led to the formation of the target cyclic carbonates 5a–5g in good to excellent conversion (60–97%). Substrates 4c and 4d gave moderate conversion for similar reasons as mentioned for the nickel based system. Furthermore post-reaction precipitation of the supporting electrolyte resulted in good recovery of Bu4NBr (∼65%).
|
|
|---|
| a General reaction conditions: [Cu(CH3CN)4][BF4] 9 (0.05 mmol) epoxide (2.0 mmol), stainless steel cathode, magnesium anode, Bu4NBr (4.2 mmol), CO2, 60 mA, 25 °C, 7 h, followed by: 50 °C, 12 h. b Conversion evaluated from the 1H NMR spectrum by integration of the epoxide vs. cyclic carbonate signals. c The cyclic carbonate was the only observed product by 1H NMR spectroscopy. |
|
In order to prove in situ formation of copper catalyst 10, the requirement for a catalyst and Bu4NBr addition, control experiments were carried out, involving the attempted synthesis of cyclic carbonate 5a from test substrate 4a under optimum conditions. Electrocarboxylation using copper catalyst [Cu(Br)4] 10 synthesised in CH3CN or EtOH resulted in comparable conversion (81% and 83%), to the in situ formed complex (90%).
Electrocarboxylation using less Bu4NBr (0.1 eq.) and Bu4NPF6 (0.1 eq.), resulted in decreased conversion (15%) in comparison to Bu4NBr (2.1 eq.) (90%). A repeat of this reaction in the absence of catalyst, also resulted in conversion of starting material (16%), although the conversion obtained was low, the result suggested the possibility of catalyst free electrocarboxylation in the presence of higher Bu4NBr loading and CO2 flow rates. Bu4NPF6 supporting electrolyte was selected in order to maintain a constant current during the reaction furthermore the PF6− counter-ions are labile and therefore non-coordinating. Finally electrocarboxylation in the absence of current between electrodes, resulted in no product formation.
Investigations of a catalyst free system
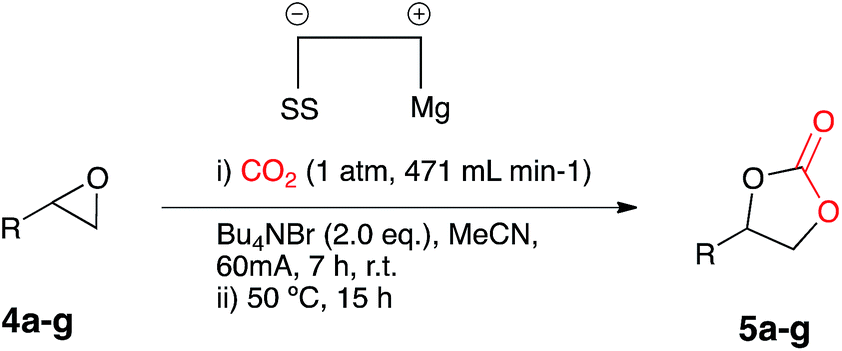
After conducting control experiments without the addition of a metal catalyst it was observed that some conversion to cyclic carbonate occurs (16%). This was particularly interesting as it opened up the possibility of direct CO2 insertion without the requirement for an additional catalyst. In order to attempt to optimise the catalyst free system an initial test reaction was carried out using styrene oxide 4a and the optimised conditions used for Cu(I) catalysis, but with an increased CO2 flow rate and the absence of a catalyst. Electrocarboxylation in acetonitrile in a single compartment cell containing a magnesium anode, stainless steel cathode and Bu4NBr (2 eq.), at constant current: 60 mA (7 h) with constant CO2 flow (471 mL min−1) followed by heating at 50 °C (12 h) formed the target cyclic carbonate 5a, in good conversion (75%) (Scheme 5).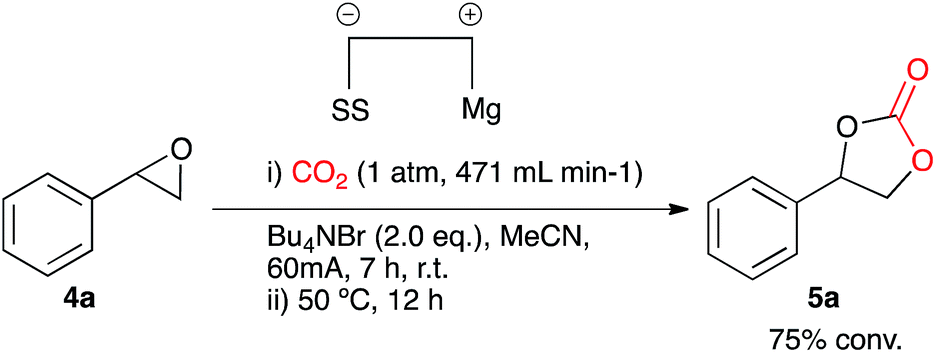 | ||
| Scheme 5 Initial catalyst free conditions. | ||
In order to determine whether the result was anomalous or restricted to aryl epoxides, a range of substrates were employed using the conditions described in Scheme 5 (Table 3). Electrocarboxylation of epoxides 4a–4g led to the formation of the target cyclic carbonates 5a–5g in poor to excellent conversion (33–91%). Again substrates 5c and 5d gave moderate conversion for similar reasons as the nickel and copper catalysed systems. In general all substrates produced lower conversions in comparison to the Cu(I) catalysed system, however, we observed that the copper catalyst 9 undergoes cathodic electroplating, thus suggesting that this system was actually a pseudo-catalyst free system, with electroplated copper appearing to enhance the conversion of the epoxide starting materials in comparison to the nickel catalysed systems. Furthermore as the results for the catalyst loading study for copper catalyst 9 were very similar, it is believed that once a layer of copper has been deposited on the cathode further plating has little effect on the reaction.
|
|
|---|
| a General reaction conditions: epoxide (2.0 mmol), stainless steel cathode, magnesium anode, Bu4NBr (4.0 mmol), CO2, 60 mA, 25 °C, 7 h, followed by: 50 °C, 12 h. b Conversion evaluated from the 1H NMR spectrum by integration of the epoxide vs. cyclic carbonate signal. c The cyclic carbonate was the only observed product by 1H NMR spectroscopy. |
|
The observed conversion of a range epoxide starting materials in the absence of a catalyst and in the presence of a magnesium anode and stainless steel cathode, suggested that the electrochemical system had the potential to be effective under catalyst free conditions. The proficiency of the process, however, was decreased because of the high CO2 flow rate being used, thus the need for a reduction in CO2 waste was investigated. A test reaction involved flushing a single compartment cell containing a magnesium anode, stainless steel, Bu4NBr supporting electrolyte (2 eq.) and test substrate 4a in acetonitrile for 1 h with CO2, followed by electrocarboxylation at a constant current (60 mA, 7 h) under a CO2 balloon with heating at 50 °C (12 h), resulting in cyclic carbonate 5a formation with good conversion (75%).
In order to determine the ideal electrode couple for the reaction system, a range of cathodic electrode materials were screened. Electrocarboxylation of 4a under the conditions described above using a copper or graphite cathode formed the corresponding cyclic carbonate 5a in excellent conversion for copper (99%), whereas graphite produced the desired product in slightly lower conversion (85%). It appeared that copper was a superior cathode in comparison to stainless steel (75%) and although graphite resulted in greater conversion of starting material, slight degradation of the electrode in acetonitrile solvent was observed.
A range of anodic electrode materials were also screened. Electrocarboxylation of 4a under the test reaction conditions using either an aluminium, zinc, tin or copper anode formed the target cyclic carbonate 5a in good conversion for aluminium (75%) and poor conversion for zinc (5%) and tin (10%), and in comparison, magnesium remained superior (99%).
Identification of a magnesium anode and a copper cathode as the optimum electrode couple, was followed by full optimisation of the catalyst free conditions. Previously it was thought that two equivalents of Bu4NBr supporting electrolyte were required in order to form stoichiometric MgBr2in situ, on the contrary, control reaction results obtained in the stainless steel cathode and magnesium anode catalyst free system suggest otherwise, therefore the Bu4NBr loading was investigated. Electrocarboxylation of 4a under the test reaction conditions, conversely, in the presence of a copper cathode and supporting electrolyte Bu4NBr at 50, 100 and 200 mol% loading, produced the cyclic carbonate 5a, with conversion increasing with electrolyte loading. Although 200 mol%, gave the highest conversion of starting material (99%), 100 mol% gave an almost equal conversion (98%), thus 100 mol% was selected as the optimum. The requirement of heating was also investigated with electrocarboxylation of 4a under the test reaction conditions in the presence of a copper cathode, Bu4NBr (100 mol%) and the absence of a heating step, resulting in target product 5a formation in good conversion (72%) with an increase in the electrolysis time to 10 h again in the absence of a heating step, resulting in only moderately higher conversion (77%). In order to decrease the overall reaction time and energy requirement, electrocarboxylation of 4a was carried out under the test reaction conditions in the presence of a copper cathode and Bu4NBr (100 mol%) with simultaneous heating, the reaction proceeded to completion in 6 h giving excellent conversion to the corresponding cyclic carbonate 5a (98%), with a decrease in reaction time to 3 h leading to moderate conversion (76%). The final optimisation reaction involved the electrocarboxylation (6 h) of 4a in air under the test reaction conditions in the presence of a copper cathode and Bu4NBr (100 mol%) with simultaneous heating, however, this resulted in the return of starting material.
The optimum applied current was also investigated and electrocarboxylation of 4a under the test reaction conditions in the presence of a copper cathode, Bu4NBr (100 mol%), and simultaneous heating at an applied current of either 20, 40, 60, 80 or 100 mA, resulted in the formation of 5a, with conversion increasing with applied current. The highest conversions (100%) were observed at 60, 80 and 100 mA, whereas 20 and 40 mA gave poor to moderate conversion, as there was no need for additional current, 60 mA was maintained as the optimum applied current.
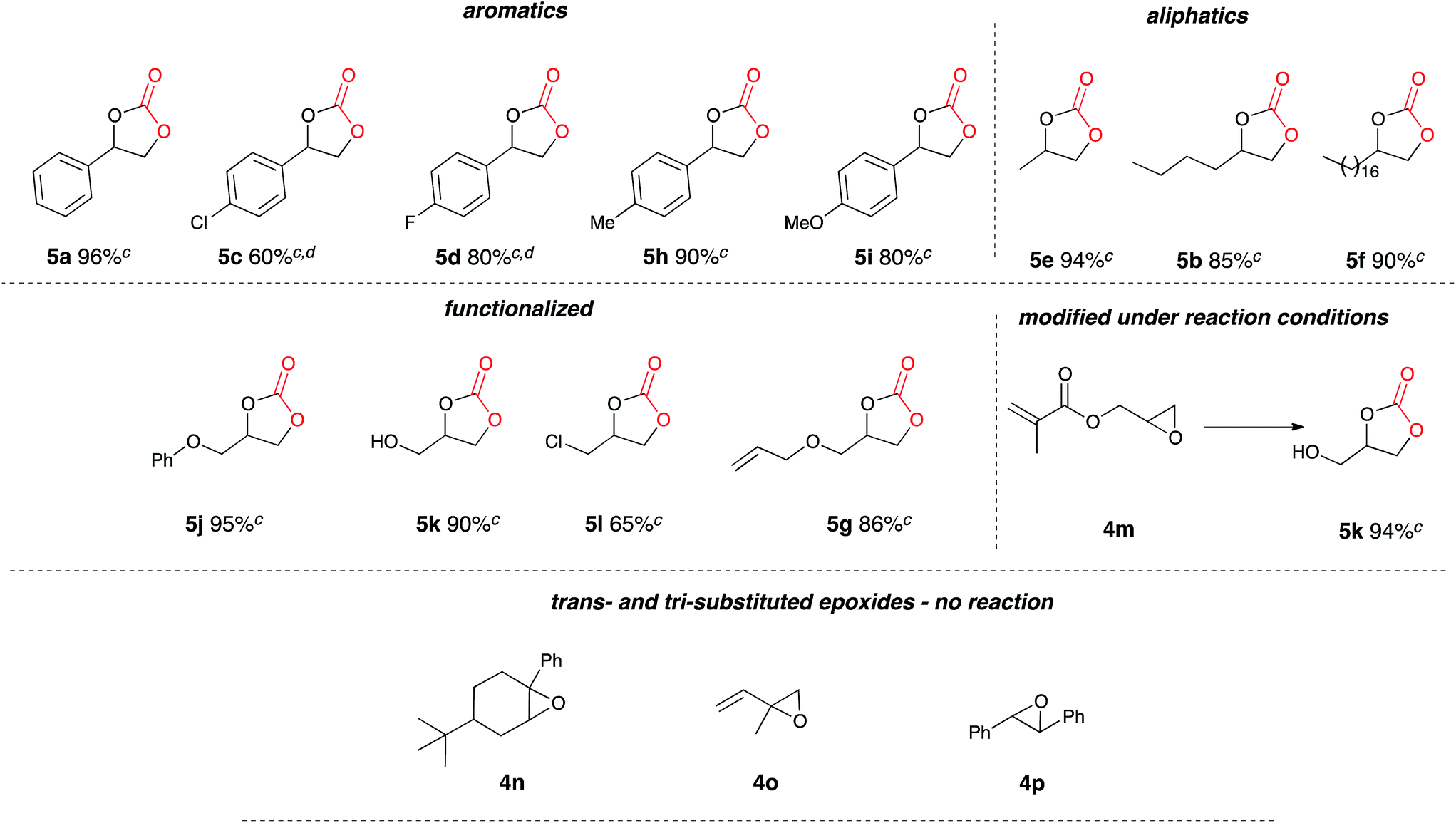
Once the optimum reaction conditions were established: sealed single compartment cell, magnesium anode, copper cathode, 100 mol% Bu4NBr loading and simultaneous heating and electrolysis at 60 mA/50 °C for 6 h under a CO2 balloon, a range of epoxide substrates were employed in order to determine the scope of the reaction conditions (Table 4). Electrocarboxylation of epoxides 4a–p led to the formation of the target cyclic carbonates 4a–l in good to excellent conversion (75–99%) and good to excellent yield (65–96%). Again substrates 4c and 4d initially gave moderate conversion attributed to the inductively electron withdrawing (−I) properties of the fluoro- and chloro- aromatic ring substituents, however an increase in the reaction time to 10 h enhanced both conversion and yield. In line with other studies in the area,7,10 epoxides 4n, 4o and 4p did not form the target cyclic carbonate product, and as the substrates are either di- or tri-substituted the lack of reactivity is attributed to steric hinderance, thus preventing the addition of Br− to ring open the epoxide, and hence reactions resulted in the return of starting material. Interestingly epoxide 4m was hydrolysed and converted into cyclic carbonate 5j. Furthermore, post-reaction precipitation of the supporting electrolyte from these reactions resulted in good recovery of Bu4NBr (∼90%).
|
|
|---|
| a General conditions: CO2, Cu cathode, Mg anode, Bu4NBr (2.0 equiv.), MeCN, single compartment cell, 60 mA, 6 h 50 °C. b On average 90–95% of the Bu4NBr is recovered after each reaction by precipitation with EtOAc and the cyclic carbonate was the only observed product by 1H NMR spectroscopy. c Isolated yield after column chromatography. d Reaction ran for 8 h. |
|
In order to explore the mild nature of these reaction conditions we were interested in the applicability of this system to enantiopure epoxides (Scheme 6). Application of our optimized conditions to the CO2 incorporation of (S)-styrene oxide (S)-4a afforded the cyclic carbonate (S)-5a in excellent yield (97%) with retention of configuration and only a slight loss of optical purity (99.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 e.r. to 98.6:1.4 e.r.).
:0.5 e.r. to 98.6:1.4 e.r.).
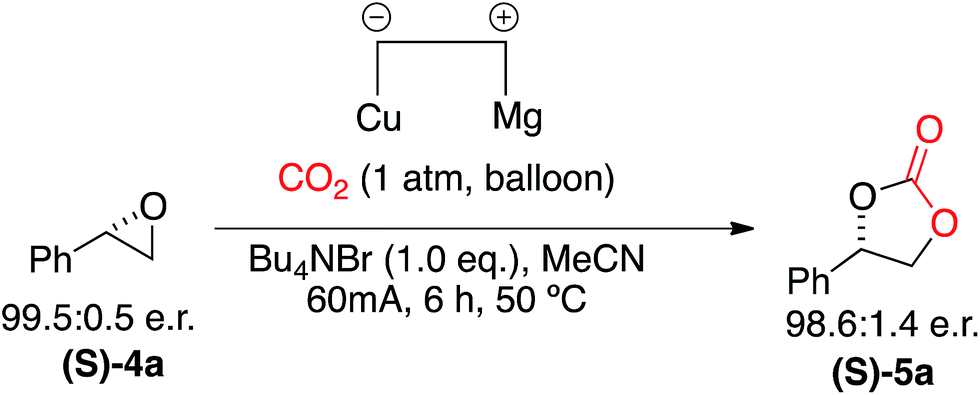 | ||
| Scheme 6 Electrocarboxylation of (S)-styrene oxide (S)-4a to afford the enantiomerically pure cyclic carbonate (S)-5a with retention of configuration. | ||
The scalability of the reaction for large scale synthesis was explored using propylene oxide 4g as the test substrate (Table 4), primarily due to the carboxylated product: 4-methyl-1,3-dioxolan-2-one (propylene carbonate) 5g being currently synthesised in industry for use as a polar aprotic solvent. Electrocarboxylation of 4g (170 mmol) under optimised conditions resulted in the formation of the target product 5g in excellent conversion (100%, 91% yield).
A proposed mechanism of our process is highlighted in Scheme 7. Under standard heating and stirring conditions we believe the pathway shown is in operation due to the recent reports by North and coworkers7e and the retention of stereochemical information in the final cyclic carbonate product.
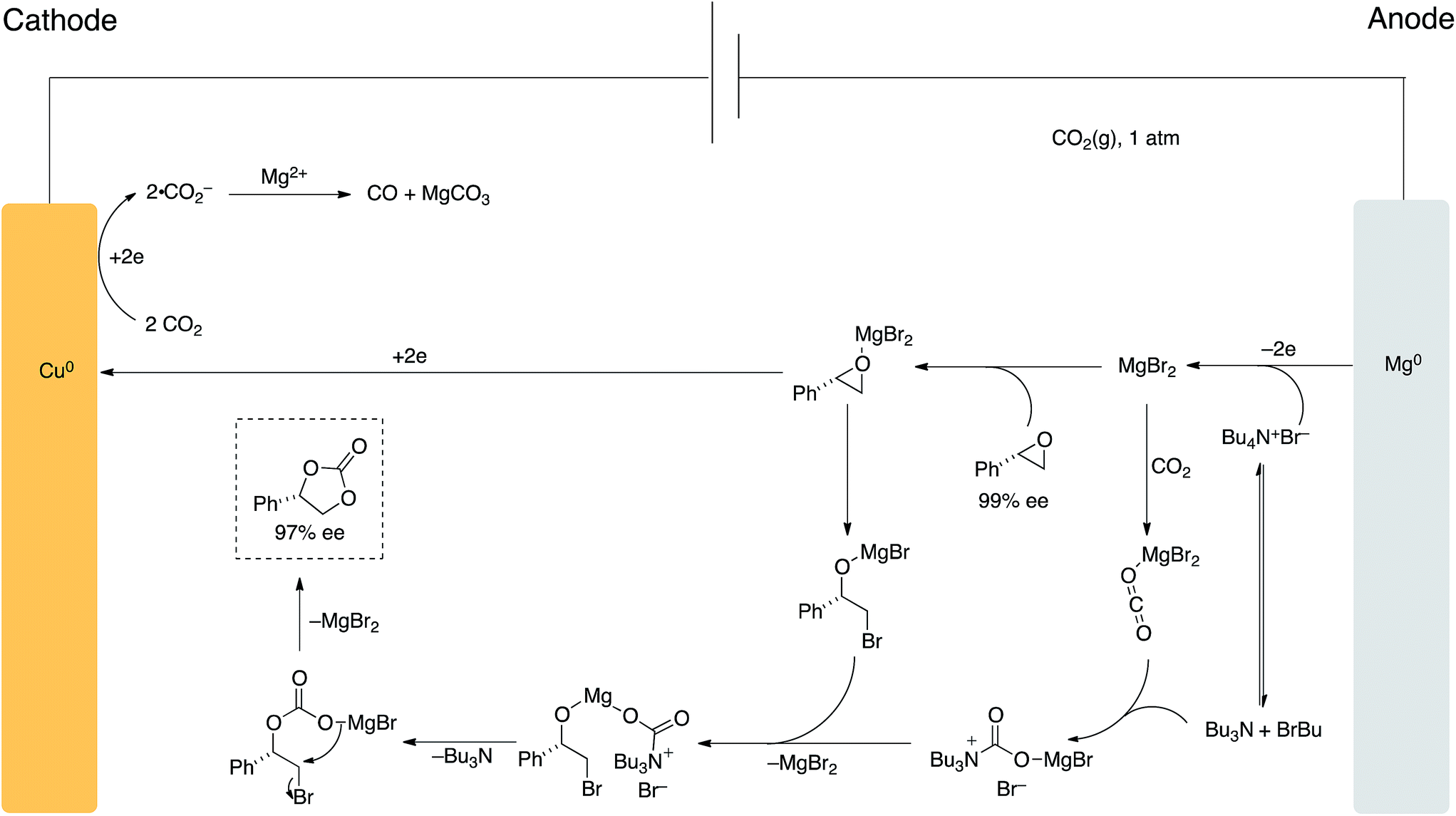 | ||
| Scheme 7 Proposed mechanism for carbon dioxide ‘ring-expansion addition’ of epoxides using applied potential. | ||
Conclusions
In conclusion we have studied the effect of several transition metal catalysts and electrode materials on the ring-expansion addition of epoxides with carbon dioxide. This process is one of a handful of emerging approaches that allow this reaction to proceed at atmospheric pressure and at ambient-mild temperatures. The yields obtained are comparable or better than those already reported in the area and our approach benefits from not employing an additional catalyst into the reaction, which in some cases can be toxic or expensive. The equipment required to perform this CO2 incorporation reaction is cheap and should be readily available in any undergraduate teaching facility i.e. copper wire, magnesium ribbon and a power supply.Experimental
Representative procedure for the ring expansion addition reaction using nickel based catalysts (Scheme 2)
Styrene oxide 4a (0.24 g, 2.0 mmol) in acetonitrile (50 mL) was electrolysed at constant current: 60 mA for 7 h in a single compartment cell containing a stainless steel cathode and magnesium anode, in the presence of catalyst (10 mol%), supporting electrolyte: Bu4NBr (01.28 g, 4 mmol) and CO2, followed by heating at 50 °C for 12 h or 18 h. On completion the reaction mixture was filtered and the filtrate washed with HCl (50 mL/0.1 M) followed by extraction using Et2O (3 × 35 mL). The combined organic extracts were washed with H2O (50 mL), dried over MgSO4 and evaporated to dryness affording 4-phenyl-1,3-dioxolan-2-one 5b as a crude amber oil. [Ni(bipy)3][(BF4)2] 2 (0.10 g, 10 mol%) (conv.: 65%); Ni[(phen)3][(BF4)2] 3 (0.12 g, 10 mol%) (conv.: 53%).Catalyst loading study using 7 and 4b
1,2-Epoxyoctadecane 4b (0.54 g, 2.0 mmol) in acetonitrile (50 mL) was electrolysed at constant current: 60 mA with constant stirring for 7 h in a single compartment cell containing a stainless steel cathode and magnesium anode, in the presence of [Ni(II)][(BF4)2] 7, supporting electrolyte: Bu4NBr and CO2, followed by heating at 50 °C for 15 h. On completion the reaction mixture was filtered and the filtrate washed with HCl (50 mL/0.1 M) followed by extraction using Et2O (3 × 35 mL). The combined organic extracts were washed with H2O (50 mL), dried over MgSO4 and evaporated to dryness affording 4-hexadecyl-1,3-dioxolan-2-one 5b as a brown solid. [Ni(II)][(BF4)2] (0.068 g, 10 mol%) and Bu4NBr (1.55 g, 4.8 mmol) (conv: 53%). [Ni(II)][(BF4)2] (0.10 g, 15 mol%) and Bu4NBr (1.68 g, 5.2.0 mmol) (conv: 66%). [Ni(II)][(BF4)2] (0.14 g, 20 mol%) and Bu4NBr (1.81 g, 5.6 mmol) (conv: 73%). [Ni(II)][(BF4)2] (0.34 g, 50 mol%) and Bu4NBr (2.56 g, 8 mmol) (conv: 90%).Representative procedure for the ring expansion addition reaction using [Ni(II)][(BF4)2] 7 (Table 1)
Styrene oxide 4a (2.0 mmol, 0.24 g) in acetonitrile (50 mL) was electrolysed at constant current: 60 mA with constant stirring for 7 h in a single compartment cell containing a stainless steel cathode and magnesium anode, in the presence of [Ni(II)][(BF4)2] 7 (0.34 g, 50 mol%), supporting electrolyte: Bu4NBr (2.56 g, 8 mmol) and CO2 (flow rate: 471 mL min−1), followed by heating at 50 °C for 15 h. On completion the reaction mixture was filtered and the filtrate washed with HCl (50 mL/0.1 M) followed by extraction using Et2O (3 × 35 mL). The combined organic extracts were washed with H2O (50 mL), dried over MgSO4 and evaporated to dryness, followed by purification by column chromatography on silica affording 4-phenyl-1,3-dioxolan-2-one 5a as a colourless solid (90% conv.).Representative procedure for the ring expansion addition reaction using [Cu(MeCN)4 (BF4)] 9 (Table 2)
Styrene oxide 4a (0.24 g, 2.0 mmol) in acetonitrile (150 mL) was electrolysed at constant current: 60 mA with constant stirring for 7 h in a single compartment cell containing a stainless steel cathode and magnesium anode, in the presence of [Cu(CH3CN)4][BF4] 9 (0.018 g, 2.5 mol%), supporting electrolyte: Bu4NBr (1.35 g, 4.2.0 mmol) and CO2 (flow rate: 132 mL min−1), followed by heating at 50 °C for 12 h. On completion the reaction mixture was filtered and filtrate was evaporated to dryness followed by addition of EtOAc (50 mL). After 1 h precipitated Bu4NBr (0.48 g, 65% recovered) was removed by filtration and the solvent evaporated, followed by purification by column chromatography on silica affording 4-phenyl-1,3-dioxolan-2-one 5a, as a colourless solid (conv: 85% conv.).Representative procedure for the ring expansion addition reaction using a stainless steel anode and magnesium cathode under catalyst free conditions (Table 3)
Styrene oxide 4a (0.24 g, 2.0 mmol) in acetonitrile (150 mL) was electrolysed at constant current: 60 mA with constant stirring for 7 h in a single compartment cell containing a stainless steel cathode and magnesium anode, in the presence of supporting electrolyte: Bu4NBr (1.28 g, 4 mmol) and CO2 (flow rate: 471 mL min−1), followed by heating at 50 °C for 12 h. On completion the reaction mixture was filtered and filtrate was evaporated to dryness followed by addition of EtOAc (50 mL). After 1 h precipitated Bu4NBr (∼70% recovered) was removed by filtration and the solvent evaporated, affording 4-phenyl-1,3-dioxolan-2-one 5a, as a crude amber oil (conv: 75%).General Procedure for the ring expansion addition reaction using a copper anode and magnesium cathode under catalyst free conditions (Table 4)
Representative procedure for the synthesis of phenyl ethylene carbonate 5a: Styrene oxide 4a (0.12 g, 1.0 mmol) and CO2 (balloon) in acetonitrile (150 mL) were electrolysed (constant current: 60 mA) for 6 h in a single compartment cell (Mg anode and copper(0) cathode) containing Bu4NBr (0.32 g, 1.0 mmol) as supporting electrolyte at 50 °C. On completion the reaction mixture was washed with aqueous 0.1 M HCl (50 mL) followed by extraction with Et2O (3 × 35 mL). The combined organic extracts were then dried over MgSO4 and evaporated under reduced pressure to afford a amber oil, which was suspended in EtOAc (100 mL). After 1 h the precipitated Bu4NBr (0.30 g, 95%) was removed by filtration and the solvent evaporated under reduced pressure to afford an amber oil. This crude material was purified by column chromatography on silica gel eluting with ethyl acetate:light petroleum. Colourless solid (0.157 g, 96%).Data for the cyclic carbonates 5a–5l
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (400 MHz, CDCl3) δ: 3.45 (1H, dd, J = 7.93, J = 7.93), 4.80 (1H, dd, J = 8.49, J = 8.33), 5.68 (1H, dd, J = 7.99, J = 7.99), 7.35–7.45 (5H, m); 13C NMR (400 MHz, CDCl3) δ: 71.2, 78.00, 125.9, 129.3, 129.8, 135.8, 154.8.
O); 1H NMR (400 MHz, CDCl3) δ: 0.93 (3H, t, J = 7.36, J = 7.38), 1.34–1.49 (2H, m), 1.65–1.74 (2H, m), 1.78–1.87 (2H, m), 4.07 (1H, dd, J = 7.2, J = 7.2), 4.53 (1H, dd, J = 7.98, J = 8.26), 4.69–4.72 (1H, m); 13C NMR (400 MHz, CDCl3) δ: 13.8, 22.8, 26.5, 29.7, 33.6, 69.4, 155.1.
O); 1H NMR (400 MHz, CDCl3) δ: 4.31 (1H, dd, J = 7.85, J = 7.89), 4.80 (1H, dd, J = 8.43, J = 8.43), 5.66 (1H, dd, J = 7.96, J = 7.96), 7.27–7.32 (2H, m), 7.42–7.44 (2H, m); 13C NMR (400 MHz, CDCl3) δ: 71.0, 77.2, 127.3, 129.5, 134.3, 135.8, 154.5.
O); 1H NMR (400 MHz, CDCl3) δ: 4.33 (1H, dd, J = 7.93, J = 8.75), 4.80 (1H, dd, J = 8.21, J = 8.59), 5.66 (1H, dd, J = 8.02, J = 8.02), 7.11–7.17 (2H, m), 7.34–7.39 (2H, m); 13C NMR (400 MHz, CDCl3) δ: 71.1, 76.7, 77.4, 116.0, 127.4, 131.5, 154.6, 162.2, 164.6.
O); 1H NMR (400 MHz, CDCl3) δ: 1.49 (3H, d, J = 6.27), 4.04 (1H, dd, J = 7.27, J = 7.27), 4.57 (1H, dd, J = 7.71, J = 7.81), 4.83–4.90 (1H, m); 13C NMR (400 MHz, CDCl3) δ: 19.4, 70.7, 73.6, 155.1.
O); 1H NMR (400 MHz, CDCl3) δ: 0.88 (3H, t, J = 7.06, J = 6.97), 1.21–1.48 (26H, m), 1.63–1.72 (2H, m), 1.77–1.86 (2H, m), 1.63–1.72 (2H, m), 4.07 (1H, dd, J = 7.43, J = 7.37), 4.52 (1H, dd, J = 8.07, J = 8.15), 4.69 (1H, m); 13C NMR (400 MHz, CDCl3) δ: 14.1, 22.7, 24.4, 29.2, 29.4, 29.4, 29.5, 29.6, 29.6, 29.7, 29.7, 31.9, 33.9, 69.4, 155.1.
C), 1792 (CO); 1H NMR (400 MHz, CDCl3) δ: 3.60–3.73 (2H, m), 4.00–4.08 (2H, m), 4.39–4.44 (1H, m), 4.50–4.57 (1H, m), 4.81–4.87 (1H, m), 5.21–5.31 (2H, m) 5.83–5.94 (1H, m); 13C NMR (400 MHz, CDCl3) δ: 66.3, 68.9, 72.5, 75.1, 117.9, 133.7, 155.1.
C), 1737 (CO); δH (400 MHz, CDCl3, Me4Si) 2.35 (3H, s), 4.34 (1H, dd, J 8.18), 4.77 (1H, dd, J 8.56), 5.64 (1H, dd, J 8.06), 7.23–7.28 (4H, m); δC (100 MHz, CDCl3, Me4Si) 21.3, 71.2, 76.9, 78.1, 126.0, 129.9, 132.7, 139.9.
O); 1H NMR (400 MHz, CDCl3) δ: 3.83 (3H, s), 4.35 (1H, dd, J = 8.14, J = 8.17), 4.75 (1H, dd, J = 8.17, J = 8.17), 5.62 (1H, dd, J = 8.05, J = 8.11), 6.94–6.97 (2H, dd, m), 7.26–7.32 (2H, m). 13C NMR (400 MHz, CDCl3) δ: 55.4, 71.1, 78.1, 114.6, 127.6, 154.9, 160.8.
O); 1H NMR (400 MHz, CDCl3) δ: 4.14–4.26 (2H, m), 4.52–4.64 (2H, m), 5.00–5.06 (1H, m), 6.90–6.92 (2H, m), 7.00–7.04 (1H, m), 7.26–7.33 (2H, m); 13C NMR (400 MHz, CDCl3) δ: 66.3, 66.9, 74.1, 114.6, 122.0, 129.7, 154.6, 157.7.
O), 3411 (O–H); 1H NMR (400 MHz, CDCl3) δ: 2.89 (1H, s), 3.71 (1H, dd, J = 3.49, J = 3.41), 3.99 (1H, dd, J = 2.97, J = 2.97), 4.50–4.56 (2H, m), 4.97–4.84 (1H, m); 13C NMR (400 MHz, CDCl3) δ: 61.7, 65.8, 76.7, 155.5.
O); 1H NMR (400 MHz, CDCl3) δ: 3.72–3.81 (2H, m), 4.42 (1H, dd, J = 5.77, J = 5.68), 4.60 (1H, dd, J = 8.30, J = 8.79), 4.94–5.00 (1H, m); 13C NMR (400 MHz, CDCl3) δ: 43.6, 66.0, 74.2, 154.1.
O); 1H NMR (400 MHz, CDCl3) δ: 3.45 (1H, dd, J = 7.93, J = 7.93), 4.80 (1H, dd, J = 8.49, J = 8.33), 5.68 (1H, dd, J = 7.99, J = 7.99), 7.35–7.45 (5H, m); 13C NMR (400 MHz, CDCl3) δ: 71.2, 78.00, 125.9, 129.3, 129.8, 135.8, 154.8.
O); 1H NMR (400 MHz, CDCl3) δ: 0.93 (3H, t, J = 7.36, J = 7.38), 1.34–1.49 (2H, m), 1.65–1.74 (2H, m), 1.78–1.87 (2H, m), 4.07 (1H, dd, J = 7.2, J = 7.2), 4.53 (1H, dd, J = 7.98, J = 8.26), 4.69–4.72 (1H, m); 13C NMR (400 MHz, CDCl3) δ: 13.8, 22.8, 26.5, 29.7, 33.6, 69.4, 155.1.
O); 1H NMR (400 MHz, CDCl3) δ: 4.31 (1H, dd, J = 7.85, J = 7.89), 4.80 (1H, dd, J = 8.43, J = 8.43), 5.66 (1H, dd, J = 7.96, J = 7.96), 7.27–7.32 (2H, m), 7.42–7.44 (2H, m); 13C NMR (400 MHz, CDCl3) δ: 71.0, 77.2, 127.3, 129.5, 134.3, 135.8, 154.5.
O); 1H NMR (400 MHz, CDCl3) δ: 4.33 (1H, dd, J = 7.93, J = 8.75), 4.80 (1H, dd, J = 8.21, J = 8.59), 5.66 (1H, dd, J = 8.02, J = 8.02), 7.11–7.17 (2H, m), 7.34–7.39 (2H, m); 13C NMR (400 MHz, CDCl3) δ: 71.1, 76.7, 77.4, 116.0, 127.4, 131.5, 154.6, 162.2, 164.6.
O); 1H NMR (400 MHz, CDCl3) δ: 1.49 (3H, d, J = 6.27), 4.04 (1H, dd, J = 7.27, J = 7.27), 4.57 (1H, dd, J = 7.71, J = 7.81), 4.83–4.90 (1H, m); 13C NMR (400 MHz, CDCl3) δ: 19.4, 70.7, 73.6, 155.1.
O); 1H NMR (400 MHz, CDCl3) δ: 0.88 (3H, t, J = 7.06, J = 6.97), 1.21–1.48 (26H, m), 1.63–1.72 (2H, m), 1.77–1.86 (2H, m), 1.63–1.72 (2H, m), 4.07 (1H, dd, J = 7.43, J = 7.37), 4.52 (1H, dd, J = 8.07, J = 8.15), 4.69 (1H, m); 13C NMR (400 MHz, CDCl3) δ: 14.1, 22.7, 24.4, 29.2, 29.4, 29.4, 29.5, 29.6, 29.6, 29.7, 29.7, 31.9, 33.9, 69.4, 155.1.
C), 1792 (CO); 1H NMR (400 MHz, CDCl3) δ: 3.60–3.73 (2H, m), 4.00–4.08 (2H, m), 4.39–4.44 (1H, m), 4.50–4.57 (1H, m), 4.81–4.87 (1H, m), 5.21–5.31 (2H, m) 5.83–5.94 (1H, m); 13C NMR (400 MHz, CDCl3) δ: 66.3, 68.9, 72.5, 75.1, 117.9, 133.7, 155.1.
C), 1737 (CO); δH (400 MHz, CDCl3, Me4Si) 2.35 (3H, s), 4.34 (1H, dd, J 8.18), 4.77 (1H, dd, J 8.56), 5.64 (1H, dd, J 8.06), 7.23–7.28 (4H, m); δC (100 MHz, CDCl3, Me4Si) 21.3, 71.2, 76.9, 78.1, 126.0, 129.9, 132.7, 139.9.
O); 1H NMR (400 MHz, CDCl3) δ: 3.83 (3H, s), 4.35 (1H, dd, J = 8.14, J = 8.17), 4.75 (1H, dd, J = 8.17, J = 8.17), 5.62 (1H, dd, J = 8.05, J = 8.11), 6.94–6.97 (2H, dd, m), 7.26–7.32 (2H, m). 13C NMR (400 MHz, CDCl3) δ: 55.4, 71.1, 78.1, 114.6, 127.6, 154.9, 160.8.
O); 1H NMR (400 MHz, CDCl3) δ: 4.14–4.26 (2H, m), 4.52–4.64 (2H, m), 5.00–5.06 (1H, m), 6.90–6.92 (2H, m), 7.00–7.04 (1H, m), 7.26–7.33 (2H, m); 13C NMR (400 MHz, CDCl3) δ: 66.3, 66.9, 74.1, 114.6, 122.0, 129.7, 154.6, 157.7.
O), 3411 (O–H); 1H NMR (400 MHz, CDCl3) δ: 2.89 (1H, s), 3.71 (1H, dd, J = 3.49, J = 3.41), 3.99 (1H, dd, J = 2.97, J = 2.97), 4.50–4.56 (2H, m), 4.97–4.84 (1H, m); 13C NMR (400 MHz, CDCl3) δ: 61.7, 65.8, 76.7, 155.5.
O); 1H NMR (400 MHz, CDCl3) δ: 3.72–3.81 (2H, m), 4.42 (1H, dd, J = 5.77, J = 5.68), 4.60 (1H, dd, J = 8.30, J = 8.79), 4.94–5.00 (1H, m); 13C NMR (400 MHz, CDCl3) δ: 43.6, 66.0, 74.2, 154.1.
Acknowledgements
B.R.B. and K.G.U.W. would like to thank Research Councils UK for RCUK fellowships and Loughborough University for funding a PhD studentship (to A.P.P.).Notes and references
- R. J. Pearson, et al. , Proc. IEEE, 2012, 100, 440 CrossRef CAS.
- Contribution of Working Group III to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, ed. B. Metz, O.R. Davidson, P.R. Bosch, R. Dave and L.A. Meyer, Cambridge University Press, United Kingdom and New York, NY, USA., 2007 Search PubMed.
- P. Styring, D. Jansen, H. de Coninck and K. Armstrong, Carbon Capture and Utilisation in the green economy, Report No. 501, The Centre For Low Carbon Futures, July 2011, ISBN 978-0-9572588-0-8 Search PubMed.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC.
- For some recent reviews and articles on high pressure and/or temperature approaches to cyclic carbonates see: (a) C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482 RSC; (b) N. Kielland, C. J. Whiteoak and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2115 CrossRef CAS; (c) I. Omae, Coord. Chem. Rev., 2012, 256, 1384 CrossRef CAS PubMed; (d) D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665–2671 RSC; (e) S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460 CrossRef CAS PubMed; (f) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC; (g) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388 CrossRef CAS PubMed.
- (a) T. Ema, Y. Miyazaki, T. Taniguchi and J. Takada, Green Chem., 2013, 15, 2485 RSC; (b) T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489 RSC; (c) Z.-Z. Yang, Y.-N. Zhao, L.-N. He, J. Gao and Z.-S. Yin, Green Chem., 2012, 14, 519 RSC; (d) D. Bai, Q. Wang, Y. Song, B. Li and H. Jing, Catal. Commun., 2011, 12, 684 CrossRef CAS PubMed; (e) F. Ahmadi, S. Tangestaninejad, M. Moghadam, V. Mirkhani, I. Mohammadpoor-Baltork and A. R. Khosropour, Inorg. Chem. Commun., 2011, 14, 1489 CrossRef CAS PubMed; (f) S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. Han, Chem. Commun., 2011, 47, 2131 RSC; (g) L. Han, H.-J. Choi, S.-J. Choi, B. Liu and D.-W. Park, Green Chem., 2011, 13, 1023 RSC; (h) H. Zhou, Y.-M. Wang, W.-Z. Zhang, J.-P. Qu and X.-B. Lu, Green Chem., 2011, 13, 644 RSC; (i) Y. Tsutsumi, K. Yamakawa, M. Yoshida, T. Ema and T. Sakai, Org. Lett., 2010, 12, 5728 CrossRef CAS PubMed; (j) M. Ulusoy, E. Çetinkaya and B. Çetinkaya, Appl. Organomet. Chem., 2009, 23, 68 CrossRef CAS; (k) J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, Green Chem., 2009, 11, 1031 RSC.
- (a) M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676 CrossRef CAS PubMed; (b) A. Buchard, F. Jutz, M. R. Kember, A. J. White, H. S. Rzepa and C. K. Williams, Macromolecules, 2012, 45, 6781 CrossRef CAS; (c) M. R. Kember, F. Jutz, A. Buchard, A. J. P. White and C. K. Williams, Chem. Sci., 2012, 3, 1245 RSC; (d) J. G. Kim, C. D. Cowman, A. M. LaPointe, U. Wiesner and G. W. Coates, Macromolecules, 2011, 44, 1110 CrossRef CAS; (e) F. Jutz, A. Buchard, M. R. Kember, S. B. Fredriksen and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395 CrossRef CAS PubMed; (f) G.-P. Wu, S.-H. Wei, W.-M. Ren, X.-B. Lu, T.-Q. Xu and D. J. Darensbourg, J. Am. Chem. Soc., 2011, 133, 15191 CrossRef CAS PubMed; (g) H. Li and Y. Niu, Appl. Organomet. Chem., 2011, 25, 424 CrossRef CAS; (h) K. Nakano, K. Kobayashi and K. Nozaki, J. Am. Chem. Soc., 2011, 133, 10720 CrossRef CAS PubMed; (i) A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2010, 47, 212 RSC; (j) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2010, 47, 141 RSC; (k) K. Nakano, S. Hashimoto and K. Nozaki, Chem. Sci., 2010, 1, 369 RSC; (l) D. J. Darensbourg, Inorg. Chem., 2010, 49, 10765 CrossRef CAS PubMed; (m) J. Yoo, S. J. Na, H. C. Park, A. Cyriac and B. Y. Lee, Dalton Trans., 2010, 2622 RSC; (n) S. I. Vagin, R. Reichardt, S. Klaus and B. Rieger, J. Am. Chem. Soc., 2010, 132, 14367 CrossRef CAS PubMed; (o) M. R. Kember, A. J. P. White and C. K. Williams, Inorg. Chem., 2009, 48, 9535 CrossRef CAS PubMed; (p) M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 48, 931 CrossRef CAS PubMed; (q) K. Nakano, M. Nakamura and K. Nozaki, Macromolecules, 2009, 42, 6972 CrossRef CAS.
- (a) M. North, B. Wang and C. Young, Energy Environ. Sci., 2011, 4, 4163 RSC; (b) M. North and C. Young, Catal. Sci. Technol., 2011, 1, 93 RSC; (c) W. Clegg, R. W. Harrington, M. North and P. Villuendas, J. Org. Chem., 2010, 75, 6201 CrossRef CAS PubMed; (d) W. Clegg, R. W. Harrington, M. North, F. Pizzato and P. Villuendas, Tetrahedron: Asymmetry, 2010, 21, 1262 CrossRef CAS PubMed; (e) W. Clegg, R. W. Harrington, M. North and R. Pasquale, Chem.–Eur. J., 2010, 16, 6828 CrossRef CAS PubMed; (f) I. S. Metcalfe, M. North, R. Pasquale and A. Thursfield, Energy Environ. Sci., 2010, 3, 212 RSC; (g) M. North, P. Villuendas and C. Young, Chem.–Eur. J., 2009, 15, 11454 CrossRef CAS PubMed; (h) M. North, Angew. Chem., Int. Ed., 2009, 48, 4104 CrossRef CAS PubMed; (i) M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946 CrossRef CAS PubMed; (j) J. Meléndez, M. North and P. Villuendas, Chem. Commun., 2009, 2577 RSC; (k) J. Meléndez, M. North and R. Pasquale, Eur. J. Inorg. Chem., 2007, 3323 CrossRef.
- (a) F. Castro-Gómez, G. Salassa, A. W. Kleij and C. Bo, Chem.–Eur. J., 2013, 19, 6289 CrossRef PubMed; (b) C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adán, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1288 CrossRef PubMed; (c) C. J. Whiteoak and A. W. Kleij, Synlett, 2013, 1748 CAS; (d) C. J. Whiteoak, E. Martin, M. M. Belmonte, J. Benet-Buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469 CrossRef CAS; (e) A. Coletti, C. J. Whiteoak, V. Conte and A. W. Kleij, ChemCatChem, 2012, 4, 1190 CrossRef CAS.
- (a) P. Tascedda and E. Duñach, J. Chem. Soc., Chem. Commun., 1995, 43–44 RSC; (b) E. Duñach, P. Tascedda, M. Weidmann and E. Dinjus, Appl. Organomet. Chem., 2001, 15, 141 CrossRef.
- (a) B. R. Buckley, A. P. Patel and K. G. U. Wijayantha, Chem. Commun., 2011, 47, 11888 RSC; (b) B. R. Buckley, Y. Chan, N. Dreyfus, C. Elliott, F. Marken and P. C. B. Page, Green Chem., 2012, 14, 2221 RSC; (c) P. C. B. Page, F. Marken, C. Williamson, Y. Chan, B. R. Buckley and D. Bethell, Adv. Synth. Catal., 2008, 350, 1149 CrossRef CAS.
- (a) A. A. Tahir and K. G. U. Wijayantha, J. Photochem. Photobiol., A, 2010, 216, 119 CrossRef CAS PubMed; (b) A. A. Tahir, K. G. U. Wijayantha, S. Saremi-Yarahmadi, M. Mazhar and V. McKee, Mater. Chem., 2009, 21, 3763 CrossRef CAS; (c) K. Tennakone, G. R. R. A. Kumara, K. G. U. Wijayantha, I. R. M. Kottegoda, V. P. S. Perera and M. L. P. Aponsu, J. Photochem. Photobiol., A, 1997, 108, 175 CrossRef CAS.
- E. Dunach and J. Perichon, J. Organomet. Chem., 1988, 352, 239 CrossRef CAS.
- (a) A.V. Getsis and Z. Mudring, Anorg. Allg. Chem., 2008, 634, 2130 CrossRef CAS; (b) A.V. Getsis and Z. Mudring, Anorg. Allg. Chem., 2008, 634, 619 CrossRef CAS.
- E. Styczen, W. K. Jozwiak, M. Gazda and D. Wyrzykowski, J. Therm. Anal. Calorim., 2008, 91, 979 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |