
Total synthesis of MECA-79†
Archanamayee Behera,
Madhu Emmadi and
Suvarn S. Kulkarni*
Department of Chemistry, Indian Institute of Technology-Bombay, Powai, Mumbai, 400076, India. E-mail: suvarn@chem.iitb.ac.in; Fax: +91-22-2576-7152; Tel: +91-22-2576-7166
First published on 29th October 2014
Abstract
The MECA-79 antigen is a sulfated mucin type core-1 extended O-glycan which is a potential anti-inflammatory agent. Herein we report a total synthesis of MECA-79 via a convergent [2 + 2] glycosylation route. The synthesis relies on efficient transformation of D-glucosamine into the orthogonally protected Tn antigen derivative and its elaboration into the TF antigen en route to MECA-79.
Introduction
Mucin type O-glycoproteins are heavily O-glycosylated proteins which are ubiquitously distributed on the surface of epithelial tissues. The O-glycans although show a great structural diversity, they all contain a common building block, comprising an N-acetyl D-galactosamine (GalNAc) residue α-linked to L-serine/L-threonine, called the Tn antigen.1 The extension of Tn antigen at O3 and O6 with D-galactose, GlcNAc and GalNAc generates various core structures (core 1 to core 8).2 Some of these O-glycans are specifically over-expressed on the surfaces of certain cancer cells and are well-established markers for tumor progression.3 The tumor associated carbohydrate antigens (TACAs), are explored for the development of anti-cancer vaccines.4–9 In 2001, Fukuda and co-workers identified a sulfated extended core-1 mucin type O-glycan called MECA-79 and proposed its structure as Galβ1 → 4(sulfo → 6)GlcNAcβ1 → 3Galβ1 → 3GalNAc 1 (Fig. 1).10 This sulfated tetrasaccharide is of significant biological interest due to its unique expression at the site of chronic inflammation. Biological studies indicate that antibodies with specificities similar to MECA-79 may serve as anti-inflammatory agents.11,12 | ||
| Fig. 1 Structure of MECA-79 antigen. | ||
Soon after its isolation, Bélot and co-workers reported a synthesis of the n-octyl derivative of MECA-79.13 Subsequently, Bertozzi and co-workers reported a synthesis of a thioether linked derivative of MECA-79 and its incorporation into a O-glycopeptide.14 They also reported the first total synthesis of L-serine linked MECA-79 en route to sulfoadhesins via regioselective glycosylations.15 In continuation of our studies directed towards the synthesis of glycosamine containing glycoconjugates,16–18 we report herein a convenient synthesis of orthogonally protected Tn antigen and its application in the total synthesis of MECA-79 antigen.
Results and discussion
A practical synthesis of an orthogonally protected Tn antigen derivative workable on a 5 g scale is shown in Scheme 1. Synthesis of Tn antigen1 entails a α-stereoselective coupling of a D-galactosamine donor with L-serine or L-threonine acceptor. Since D-galactosamine (D-GalNH2) is expensive, we selected its C4 epimer D-glucosamine (D-GlcNH2) as a cheap and abundant starting material. First, commercially available D-glucosamine was rapidly converted to fully protected derivative 2 via Hung's one-pot protection protocol in very good overall yields.19 Accordingly, starting from 5 g D-GlcNH2·HCl, the three stage sequence involving diazo transfer, per-O-silylation and one-pot protection could be carried out in 3 days to obtain 6 g of 2 after a single column purification (69% overall). Hydrolysis of the 4,6-O-benzylidene acetal of 2 followed by selective TBDPS protection of the primary hydroxyl group cleanly afforded the C4-alcohol derivative 3 (81%, over two steps). Triflation of 3 (Tf2O, pyridine) and its subsequent reaction with water in acetonitrile as a solvent under reflux conditions effected the migration of C3-OAc group to C4 position with concomitant SN2 displacement of the C4 triflate, resulting in the inversion of configuration at C4.17,20 The free C3-hydroxyl group was capped with a chloroacetyl group to furnish the desired D-galactosamine building block 4 in 89% yield over three steps. For the synthesis of Tn antigen, the anomeric acetate 4 was first treated with TMSI to generate the requisite α-glycosyl iodide, which was stereoselectively glycosylated with the known L-serine derived acceptor 5 (ref. 21) using I2 as a promoter22 to obtain the α-linked glycosyl serine derivative 6 (1H NMR H-1 δ 4.91, J = 3.6 Hz, 13C NMR C-1 δ 99.2) in 77% yield over two steps. Selective removal of the chloroacetyl group in 6 by using thiourea23 in pyridine afforded acceptor 7 (90%). The entire sequence from D-GlcNH2·HCl can be carried out in a week. | ||
| Scheme 1 Synthesis of orthogonally protected Tn antigen from D-glucosamine. | ||
The appropriately protected Tn antigen derivative 7 was utilized for the synthesis of TF antigen derivatives as shown in Scheme 2. For this purpose, the known C3-alcohol D-galactose derivative 8 (ref. 24) was first treated with chloroacetyl chloride in pyridine to obtain the fully protected D-galactose building block 9 (98%). Glycosylation of donor 9 with Tn antigen acceptor 7 under NIS, TMSOTf conditions at 0 °C afforded the corresponding TF antigen derivative 10 in 73% yields. The chloroacetyl group at O3′′ was subsequently removed with thiourea to furnish the desired C3′′-alcohol disaccharide acceptor 11 in excellent yield.
 | ||
| Scheme 2 Synthesis of TF antigen derivative 11. | ||
Scheme 3 outlines the preparation of the left hand disaccharide donor 16 and its assembly with the TF disaccharide acceptor 11 to obtain tetrasaccharide 17. For the synthesis of disaccharide 16, we started with the known 4,6-O-benzylidene protected derivative 12 (ref. 25) which was easily obtained from D-GlcNH2. By using benzoyl chloride and pyridine, the free C3-hydroxyl group in 12 was benzoylated to furnish 13, which was then subjected to a regioselective reductive ring opening of 4,6-O-benzylidene acetal by using Et3SiH and TFA26 to afford the C4 alcohol 14. Coupling of trichloroacetimidate donor 15 (ref. 27) with C4–OH acceptor 14 by using TMSOTf as a promoter at −50 °C in CH2Cl2 cleanly generated the β-linked disaccharide 16 in 60% yield along with a small amount of the corresponding aglycon transferred product (15%). Finally, thioglycoside donor 16 was coupled with acceptor 11 by using NIS, TMSOTf as a promoter to give exclusively β-linked tetrasaccharide 17 (80%) corresponding to MECA-79 antigen.
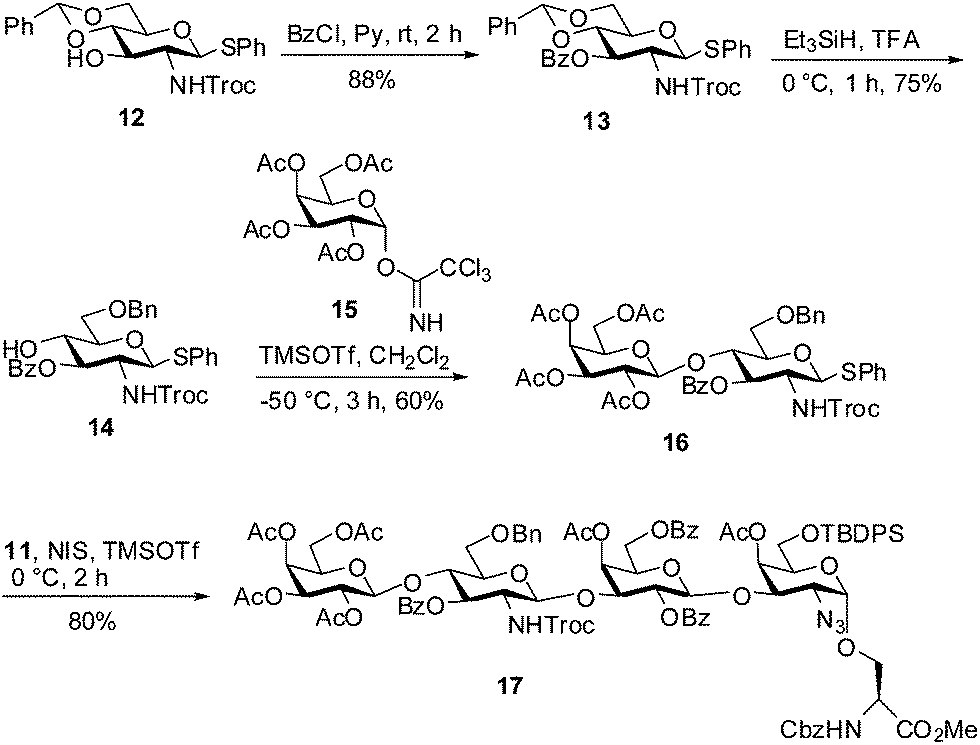 | ||
| Scheme 3 Assembly of tetrasaccharide 17. | ||
Scheme 4 outlines the global deprotection of fully protected tetrasaccharide 17. Sequential removal of TBDPS group by treating with TBAF in AcOH followed by acylation by using acetic anhydride in pyridine furnished 18 as a sole product in 87% yield over two steps. One-step reduction of both the azide and NHTroc groups by using Zn in AcOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc (2:1),28 and subsequent acetylation by treating with acetic anhydride afforded the N-acetylated derivative 19 in good yields. Oxidative debenzylation using sodium bromate and sodium dithionate29 in water and EtOAc furnished the 6′′′-OH derivative 20, which was sulfated by treating with sulfur trioxide–triethyl amine complex in pyridine as a solvent followed by removal of all the ester functionalities with NaOMe in MeOH–H2O to afford the target molecule 1 in 80% yield over two steps. The spectral data of 1 matched perfectly with the reported one confirming its identity.15 Target molecule 1 and all the synthetic intermediates are thoroughly characterized by 1H, 13C, and 2D NMR (see ESI†).
:EtOAc (2:1),28 and subsequent acetylation by treating with acetic anhydride afforded the N-acetylated derivative 19 in good yields. Oxidative debenzylation using sodium bromate and sodium dithionate29 in water and EtOAc furnished the 6′′′-OH derivative 20, which was sulfated by treating with sulfur trioxide–triethyl amine complex in pyridine as a solvent followed by removal of all the ester functionalities with NaOMe in MeOH–H2O to afford the target molecule 1 in 80% yield over two steps. The spectral data of 1 matched perfectly with the reported one confirming its identity.15 Target molecule 1 and all the synthetic intermediates are thoroughly characterized by 1H, 13C, and 2D NMR (see ESI†).
 | ||
| Scheme 4 Global deprotection and completion of the synthesis of MECA-79. | ||
Conclusions
In conclusion, we have successfully carried out the total synthesis of MECA-79 via a convergent [2 + 2] glycosylation route. The tetrasaccharide is equipped with a functional handle for further conjugation to a carrier protein or microarrays. The synthesis involves efficient transformation of D-glucosamine into the orthogonally protected Tn antigen derivative which also allows access to various mucin type oligosaccharides (core 1 to core 8) by selective removal of the protecting groups at O3 and O6 and coupling with respective glycosyl donors. Also the overall protecting group pattern of the fully protected tetrasaccharide 17 will further facilitate its elaboration into extended core 2 structures such as tetra-core 2 and hexa-core 2.Experimental section
All reactions were conducted under a dry nitrogen atmosphere. Solvents (CH2Cl2 >99%, THF 99.5%, acetonitrile 99.8%, DMF 99.5%) were purchased in capped bottles and dried under sodium or CaH2. All other solvents and reagents were used without further purification. All glassware used was oven dried before use. TLC was performed on pre-coated Aluminium plates of Silica Gel 60 F254 (0.25 mm, E. Merck). Developed TLC plates were visualized under a short-wave UV lamp and by heating plates that were dipped in ammonium molybdate/cerium(IV) sulfate solution. Silica gel column chromatography was performed using Silica gel (100–200 mesh) and employed a solvent polarity correlated with TLC mobility. NMR experiments were conducted on 500 and 400 MHz instrument using CDCl3 (D, 99.8%) or (CD3)2CO (D, 99.9%) as solvents. Chemical shifts are relative to the deuterated solvent peaks and are in parts per million (ppm). 1H-1H COSY was used to confirm proton assignments. Mass spectra were acquired in the ESI mode. Melting points were determined by capillary apparatus. Specific rotation experiments were measured at 589 nm (Na) and 20 °C. IR spectra were recorded on an FT-IR spectrometer using CsCl plates.1,3-Di-O-acetyl-2-azido-2-deoxy-6-O-t-butyldiphenylsilyl-α,β-D-glucopyranoside (3)
1,3-Di-O-acetyl-2-azido-4,6-O-benzylidene-2-deoxy-α,β-D-glucopyranoside 2 (3.50 g, 9.27 mmol) was dissolved in 80% AcOH (140 mL) and kept for reflux at 80 °C. After 1.5 h reaction mixture was concentrated in vacuo and subsequently co-evaporated with toluene (3 × 60 mL). After evaporation of solvent, the crude product was dissolved in CH3CN (35 mL) and to this solution, imidazole (1.50 g, 23.17 mmol), followed by TBDPSCl (2.85 mL, 11.12 mmol) were added. After 15 min, the reaction mixture was concentrated in vacuo and the desired product was purified by column chromatography on silica gel (10% ethyl acetate:pet ether) to afford the desired product 3 as a yellowish liquid (3.90 g, 82%, α/β = 1:8): [α]20D = +8.9 (c 1.0, CHCl3); IR (CHCl3) ν 3498, 3018, 2931, 2858, 2112, 1759, 1428, 1372, 1218, 1113, 1082, 759, 704, 506 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.67–7.64 (m, 4H, ArH), 7.45–7.35 (m, 6H, ArH), 6.24 (d, J = 3.6 Hz, H-1α), 5.53 (d, J = 8.5 Hz, H-1β), 4.97 (dd, J = 10.1, 9.4 Hz, 1H, H-3), 3.94 (dd, J = 10.9, 4.2 Hz, 1H, H-6a), 3.86 (dd, J = 10.9, 4.6 Hz, 1H, H-6b), 3.81–3.79 (m, 2H, H-4), 3.56–3.48 (m, 2H, H-2, H-5), 2.19 (s, 3H, COCH3), 2.16 (s, 3H, COCH3), 1.05 (s, 9H, (CH3)3CSi); 13C NMR (100 MHz, CDCl3) δ 171.2, 168.8, 135.8, 135.7, 132.7, 130.1, 130.09, 128.0, 127.9, 92.8, 90.4, 75.7, 75.5, 73.4, 70.5, 64.0, 62.7, 26.9, 21.1, 21.0, 19.3; HRMS-ESI [M + H]+ calcd for C26H33O7SiN3 528.2166, found 528.2161.
1,4-Di-O-acetyl-2-azido-2-deoxy-3-O-chloroacetyl-6-O-t-butyldiphenylsilyl-α,β-D-galactopyranoside (4)
Trifluoromethanesulfonic anhydride (1.07 mL, 6.37 mmol) was added dropwise at 0 °C to a stirred solution of 3 (2.8 g, 5.31 mmol), pyridine (2.56 mL, 31.8 mmol) in CH2Cl2 (40 mL). After 2 h, the reaction mixture was diluted with CH2Cl2 (100 mL) and washed successively with 1 M HCl (30 mL), aq. NaHCO3 (30 mL), and water (30 mL). Separated organic layer was dried over Na2SO4, concentrated and this crude product was used for the next step without purification.The crude product was dissolved in acetonitrile (30 mL), H2O (2.1 mL) and kept for reflux at 65 °C for 90 min. Then reaction mixture was concentrated in vacuo and the residue obtained was dissolved in EtOAc and washed with brine (3 × 50 mL). Separated organic layer was dried over Na2SO4 and concentrated. The crude product which was obtained after removal of solvent was dissolved in CH2Cl2 (30 mL). To this clear solution ClAcCl (1.3 mL, 16.0 mmol) and pyridine (1.3 mL, 16.0 mmol) were added. After 10 min, reaction mixture was concentrated in vacuo and chromatographed on silica gel (20% ethyl acetate:pet ether) to obtain the desired product 4 as a foam (2.85 g, 89%, α/β = 1:8): [α]20D = −16.8 (c 0.1, CHCl3); IR (CHCl3) ν 3685, 3020, 2400, 1520, 1424, 1216, 928, 769, 669, 627 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.60–7.56 (m, 4H, ArH), 7.46–7.35 (m, 6H, ArH), 6.27 (d, J = 3.7 Hz, H-1α), 5.58 (dd, J = 3.2, 0.9 Hz, 1H, H-4), 5.53 (d, J = 8.5 Hz, H-1β), 4.97 (dd, J = 10.8, 3.2 Hz, 1H, H-3), 4.08, 4.06 (ABq, J = 15.3 Hz, 2H, –CH2), 3.91–3.86 (m, 1H, H-6a), 3.83 (dd, J = 10.8, 8.5 Hz, 1H, H-2), 3.74 (dd, J = 9.8, 5.6 Hz, 1H, H-6b), 3.59 (dd, J = 9.8, 8.7 Hz, 1H, H-5), 2.15 (s, 3H, COCH3), 2.01 (s, 3H, COCH3), 1.01 (s, 9H, (CH3)3CSi); 13C NMR (100 MHz, CDCl3) δ 170.2, 168.7, 166.3, 135.8, 135.7, 132.7, 130.1, 128.0, 127.9, 93.0, 90.5, 73.9, 73.4, 71.2, 71.0, 65.8, 60.9, 60.5, 59.9, 57.1, 53.9, 40.6, 26.86, 26.83, 20.9, 20.7, 19.1; HRMS-ESI [M + H]+ calcd for C28H34O8SiClN3 604.1882, found 604.1870.
N-(Benzyloxycarbonyl)-O-(4-O-acetyl-3-O-chloroacetyl-2-azido-2-deoxy-6-O-t-butyldiphenylsilyl-α-D-galactopyranosyl)-L-serine methylester (6)
TMSI (0.8 mL, 5.56 mmol) was added to a clear solution of 4 (2.8 g, 4.63 mmol) in CH2Cl2 (30 mL) at 0 °C and reaction mixture was stirred at the same temperature for 2 h. After complete consumption of starting material, benzene (15 × 2 mL) was added and the mixture was azeotroped twice on a rotary evaporator under high vacuum to obtain the crude glycosyl iodide. This brownish oil was dissolved in anhydrous CH2Cl2 (30 mL) and cannulated dropwise into a mixture of acceptor (1.0 g, 3.93 mmol) and 3 Å MS (2 g) at room temperature. After 6 h the crude reaction mixture was filtered through Celite. The filtrate was concentrated and chromatographed on silica gel (20% ethyl acetate:pet ether) to obtain 6 as a viscous oily liquid (2.41 g, 77%): [α]20D = +52.6 (c 1.8, CHCl3); IR (CHCl3) ν 3018, 2929, 2113, 1749, 1216, 1046, 759, 668 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.63–7.58 (m, 5H, ArH), 7.44–7.31 (m, 10H, ArH), 5.66 (d, J = 8.4 Hz, 1H, NH), 5.59 (d, J = 3.2 Hz, 1H, H-4), 5.36 (dd, J = 11.0, 3.2 Hz, 1H, H-3), 5.09, 5.08 (ABq, J = 11.3 Hz, 2H, CH2 of Cbz), 4.91 (d, J = 3.6 Hz, 1H, H-1), 4.58–4.55 (m, 1H, –CH), 4.09–4.02 (m, 5H, H-5, –CH2, –CH2), 3.79 (s, 3H), 3.75–3.63 (m, 1H, H-6a), 3.61–3.54 (m, 2H, H-2, H-6b), 1.98 (s, 3H, CH3), 1.01 (s, 9H, (CH3)3CSi); 13C NMR (100 MHz, CDCl3) δ 170.3, 170.2, 166.4, 156.2, 136.1, 135.72, 135.71, 132.9, 132.8, 130.8, 130.08, 130.05, 128.7, 128.4, 128.3, 128.0, 99.2, 70.3, 69.5, 67.4, 67.2, 61.2, 57.6, 54.4, 53.0, 40.6, 26.8, 20.7, 19.1; HRMS-ESI [M + H]+ calcd for C38H46O11SiClN4 797.2621, found 797.2600.
N-(Benzyloxycarbonyl)-O-(4-O-acetyl-2-azido-2-deoxy-6-O-t-butyldiphenylsilyl-α-D-galactopyranosyl)-L-serine methylester (7)
Thiourea (0.36 g, 4.8 mmol) was added to a clear solution of 6 (0.54 g, 0.68 mmol) in pyridine (7 mL) and EtOH (7 mL) and the reaction mixture was kept for reflux at 80 °C. After 30 min, solvents were removed and the crude product was chromatographed on silica gel (25% ethyl acetate:pet ether) to afford 7 as a viscous liquid (0.41 g, 84%): [α]20D = +30.2 (c 1.0, CHCl3); IR (CHCl3) ν 3018, 2975, 2112, 1735, 1427, 1216, 1047, 758, 669 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.63–7.59 (m, 5H, ArH), 7.42–7.31 (m, 10H, ArH), 5.68 (d, J = 8.4 Hz, 1H, NH), 5.47 (d, J = 3.0 Hz, 1H, H-4), 5.12–5.03 (m, 2H, –CH2), 4.86 (d, J = 3.6 Hz, 1H, H-1), 4.57–4.55 (m, 1H, –CH), 4.22 (dd, J = 10.6, 3.0 Hz, 1H, H-3), 4.02–3.90 (m, 3H, H-5, –CH2), 3.78 (s, 3H, CH3), 3.74–3.58 (m, 2H, H-6a, H-6b), 3.40 (dd, J = 10.6, 3.6 Hz, 1H, H-2), 2.01 (s, 3H, CH3), 1.02 (s, 9H, (CH3)3CSi); 13C NMR (100 MHz, CDCl3) δ 171.7, 170.4, 156.1, 135.7, 133.0, 132.9, 130.1, 130.0, 128.7, 128.4, 128.3, 128.0, 99.3, 70.1, 69.7, 69.4, 67.5, 67.4, 61.4, 60.2, 54.4, 53.0, 26.8, 20.9, 19.2; HRMS-ESI [M + Na]+ calcd for C36H44O10SiN4 743.2719, found 743.2706.
Phenyl 4-acetyl-3-chloroacetyl-2,6-di-O-benzoyl-1-thio-β-D-galactopyranoside (9)
To the clear solution of 8 (2.3 g, 4.401 mmol) in CH2Cl2 (24 mL) and pyridine (1.06 mL, 13.20 mmol) was added ClAcCl (1.05 mL, 1.49 mmol) at 0 °C. Then reaction mixture was kept up to 1 h at room temperature. After consumption of the starting material, the crude reaction mixture was concentrated and chromatographed on silica gel (25% ethyl acetate:pet ether) to get 9 as a yellowish solid (2.6 g, 98%): [α]20D = +21.3 (c 1.7, CHCl3); mp 146 °C; IR (CHCl3) ν 3020, 1729, 1266, 1216, 1112, 758, 711, 668 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.02–8.00 (m, 4H, ArH), 7.62–7.44 (m, 8H, ArH), 7.26–7.23 (m, 1H, ArH), 7.19–7.15 (m, 2H, ArH), 5.60–5.54 (m, 2H, H-4, H-2), 5.36 (dd, J = 9.9, 3.3 Hz, 1H, H-3), 4.93 (d, J = 10.0 Hz, 1H, H-1), 4.55 (dd, J = 11.4, 7.2 Hz, 1H, H-6a), 4.39 (dd, J = 11.4, 5.7 Hz, 1H, H-6b), 4.19 (t, J = 6.4 Hz, 1H, H-5), 3.89–3.90 (m, 2H), 2.19 (s, 3H, COCH3); 13C NMR (125 MHz, CDCl3): δ 170.6, 166.8, 166.1, 165.4, 133.8, 133.6, 132.7, 132.6, 130.0, 129.9, 129.4, 129.2, 129.0, 128.8, 128.7, 128.3, 87.2, 74.7, 73.9, 67.8, 67.6, 62.3, 40.5, 20.8; HRMS-ESI [M + Na]+ calcd for C30H27ClO9S 621.0957, found 621.0943.
N-(Benzyloxycarbonyl)-O-(4-O-acetyl-2,6-di-O-benzoyl-3-O-chloroacetyl-β-D-galactopyranosyl-(1 → 3)-2-azido-2-deoxy-4-O-acetyl-6-O-t-butyldimethylsilyl-α-D-galactopyranosyl)-L-serine methylester (10)
NIS (0.18 g, 1.38 mmol) was added to a suspension of thiogalactoside donor 9 (0.28 g, 0.46 mmol), acceptor 7 (0.27 g, 0.37 mmol) and 3 Å MS (0.4 g) in CH2Cl2 (5 mL). Then the reaction mixture was brought to 0 °C and TMSOTf (25 μL, 0.14 mmol) was added slowly dropwise and kept stirring at rt for 2 h. After completion of the starting material, Et3N (1 mL) was added and stirred for 10 min. Then the reaction mixture was filtered through Celite, washed with aq. Na2S2O3, the separated organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel chromatography (25% ethyl acetate:pet ether) to obtain the desired product 10 as a colourless foam (0.32 g, 73%): [α]20D = +37.4 (c 0.9, CHCl3); IR (CHCl3) ν 2931, 2110, 1732, 1268, 1226, 1112, 1071, 757, 710, 614, 504 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.00–7.99 (d, J = 7.7 Hz, 4H, ArH), 7.63–7.52 (m, 6H, ArH), 7.45–7.30 (m, 15H, ArH), 5.61 (d, J = 8.6 Hz, 1H, NH), 5.53 (m, 2H, H-4, H-4′), 5.47 (dd, J = 10.4, 7.8 Hz, 1H, H-2′), 5.30 (dd, J = 10.4, 3.3 Hz, 1H, H-3′), 5.12–5.05 (m, 2H, CH2 of Cbz), 4.92 (d, J = 7.8 Hz, 1H, β, H-1′), 4.83 (d, J = 3.6 Hz, 1H, α, H-1), 4.58–4.55 (m, 2H, –CH, H-6a′), 4.33 (dd, J = 10.9, 8.1 Hz, 1H, H-6b′), 4.14 (t, J = 6.8 Hz, 1H, H-5′), 4.04 (dd, J = 10.6, 3.2 Hz, 1H, H-3), 3.95–3.91 (m, 3H, CH2, H-6a), 3.89–3.85 (m, 2H, –CH2), 3.70 (s, 3H, CO2Me), 3.63–3.54 (m, 2H, H-5, H-6b), 3.47 (dd, J = 10.6, 3.6 Hz, 1H, H-2), 2.20 (s, 3H, COCH3), 1.97 (s, 3H, COCH3), 1.02 (s, 9H, (CH3)3CSi); 13C NMR (125 MHz, CDCl3) δ 170.6, 170.4, 169.3, 166.8, 165.9, 165.2, 156.1, 136.1, 135.71, 135.70, 133.6, 133.5, 133.2, 133.1, 129.9, 129.8, 129.4, 129.2, 128.7, 128.6, 128.58, 128.5, 128.3, 127.9, 127.8, 101.7, 98.8, 74.3, 72.6, 70.8, 70.7, 69.4, 69.3, 68.9, 67.4, 66.8, 62.5, 61.1, 59.5, 54.3, 52.8, 40.5, 26.8, 20.8, 20.7, 19.2; HRMS-ESI [M + Na]+ calcd for C60H65ClN4O19Si 1231.3593 found 1231.3542.
N-(Benzyloxycarbonyl)-O-(4-O-acetyl-2,6-di-O-benzoyl-β-D-galactopyranosyl-(1 → 3)-2-azido-2-deoxy-4-O-acetyl-6-O-t-butyldimethylsilyl-α-D-galactopyranosyl)-L-serine methylester (11)
Thiourea (0.14 g, 1.90 mmol) was added to a clear solution of 10 (0.32 g, 0.27 mmol) in pyridine (3.5 mL) and EtOH (3.5 mL) and the reaction mixture was kept for reflux at 80 °C. After 30 min, solvents were removed and the crude product was chromatographed on silica gel (25% ethyl acetate:pet ether) to afford 11 as a white foam (0.28 g, 92%): [α]20D = +29.8 (c 2.8, CHCl3); IR (CHCl3) ν 3436, 3020, 2931, 2110, 1728, 1270, 1230, 1112, 1070, 758, 710, 504 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.10–8.02 (m, 4H, ArH), 7.66–7.63 (m, 5H, ArH), 7.59–7.33 (m, 16H, ArH), 5.66 (d, J = 8.0 Hz, 1H, NH), 5.56 (d, J = 2.3 Hz, 1H, H-4), 5.50 (d, J = 2.9 Hz, 1H, H-4′), 5.27–5.23 (m, 1H, H-2′), 5.14, 5.10 (ABq, J = 12.0 Hz, 2H, CH2 of Cbz), 4.90 (d, J = 7.7 Hz, 1H, β, H-1′), 4.87 (d, J = 3.5 Hz, 1H, α, H-1), 4.64–4.53 (m, 2H, –CH, H-6a′), 4.34 (dd, J = 11.1, 7.3 Hz, 1H, H-6b′), 4.09–4.05 (m, 2H, H-3, H-5′), 4.02 (dd, J = 9.8, 3.3 Hz, 1H, H-3′), 3.96–3.92 (m, 3H, CH2, H-6a), 3.74 (s, 3H, CO2Me), 3.68–3.57 (m, 2H, H-5, H-6b), 3.53 (dd, J = 10.4, 3.5 Hz, 1H, H-2), 2.22 (s, 3H, COCH3), 1.96 (s, 3H, COCH3), 1.05 (s, 9H, (CH3)3CSi); 13C NMR (125 MHz, CDCl3) δ 171.0, 170.5, 170.3, 169.2, 166.5, 166.1, 165.9, 165.2, 156.0, 136.0, 135.6, 135.5, 133.3, 133.2, 133.0, 132.9, 129.8, 129.7, 129.5, 129.4, 128.6, 128.5, 128.4, 128.3, 128.2, 128.0, 127.8, 127.74, 127.71, 101.5, 98.7, 74.5, 73.0, 72.9, 72.4, 71.1, 70.9, 70.8, 69.9, 69.5, 68.7, 67.3, 67.0, 62.5, 61.7, 59.3, 54.2, 52.7, 26.7, 20.8, 20.6, 19.1, 19.0; HRMS-ESI [M + Na]+ calcd for C58H64N4O18Si 1155.3877, found 1155.3923.
Phenyl 4,6-(benzylidene)-3-benzoyl-2-deoxy-2-[[(2,2,2-trichloroethoxy)carbonyl]amino]-1-thio-β-D-glucopyranoside (13)
BzCl (0.6 mL, 5.40 mmol), pyridine (0.43 mL, 5.40 mmol) were added to a clear solution of 12 (0.9 g, 1.80 mmol) in CH2Cl2 (10 mL) at 0 °C. The reaction mixture was stirred at room temperature for 2 h. After that the reaction mixture was diluted with CH2Cl2 and washed with NaHCO3 (3 × 20 mL) and brine (20 mL). Then dried over Na2SO4, concentrated and chromatographed on silica gel (12% ethyl acetate:pet ether) to obtain the desired product 13 as a white solid (1.0 g, 87%): [α]20D = −15.4 (c 3.3, CHCl3); mp 215 °C; IR (CHCl3) ν 3019, 1712, 1541, 1369, 1270, 1216, 1082, 1023, 759 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 7.3 Hz, 2H, ArH), 7.56–7.53 (m, 1H, ArH), 7.47–7.46 (m, 2H, ArH), 7.41–7.37 (m, 2H, ArH), 7.34–7.32 (m, 2H, ArH), 7.28–7.19 (m, 4H, ArH), 7.14–7.11 (m, 2H, ArH), 6.35 (d, J = 10.0 Hz, 1H, NH), 5.79 (t, J = 9.8 Hz, 1H, H-3), 5.46 (s, 1H, benzylidene), 4.84 (d, J = 10.4 Hz, 1H, H-1), 4.14 (d, J = 10.4 Hz, 1H, H-2), 4.63 (s, 2H, CH2 of Troc), 4.06 (dd, J = 10.2, 4.7 Hz, 1H, H-6a), 3.81 (t, J = 9.4 Hz, 1H, H-4), 3.70 (t, J = 10.2 Hz, 1H, H-6b), 3.62–3.56 (m, 1H, H-5); 13C NMR (125 MHz, CDCl3): δ 167.0, 154.6, 136.9, 133.7, 132.7, 132.5, 130.1, 129.0, 128.9, 128.5, 128.14, 128.11, 125.8, 100.9, 95.4, 88.3, 78.7, 74.4, 73.8, 70.4, 68.3, 55.5; HRMS-ESI [M + Na]+ calcd for C29H26Cl3NO7S 660.0388, found 660.0382.
Phenyl 3-benzoyl-6-benzyl-2-deoxy-2-[[(2,2,2-trichloroethoxy)carbonyl]amino]-1-thio-β-D-glucopyranoside (14)
Triethyl silane (0.5 mL, 3.0 mmol) and TFA (0.3 mL, 3.0 mmol) were added dropwise at 0 °C to a stirred solution of 13 (0.38 g, 0.60 mmol) in CH2Cl2 (4 mL) and kept the reaction mixture for 1 h. After completion of the starting material, the reaction mixture was diluted with CH2Cl2 and washed with NaHCO3 (20 mL) and brine (10 mL). Separated organic layer was dried over Na2SO4. The desired product was purified by column chromatography on silica gel by using (25% ethyl acetate:pet ether) as eluent to afford the desired product 14 as a white foam (0.29 g, 75%): [α]20D = +15.6 (c 7.6, CHCl3); IR (CHCl3) ν 3350, 3019, 2868, 1719, 1528, 1279, 1216, 1070, 1026, 820, 759, 712, 668, 570 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.93 (d, J = 7.6 Hz, 2H, ArH), 7.52–7.49 (m, 3H, ArH), 7.33–7.19 (m, 10H, ArH), 5.83–5.79 (m, 1H, NH), 5.40 (t, J = 9.6 Hz, 1H, H-3), 4.83 (d, J = 10.3 Hz, 1H, H-1), 4.67 (d, J = 12.0 Hz, 1H, CHHPh), 4.55–4.49 (m, 3H, CHHPh, CH2 of NHTroc), 4.00–3.94 (m, 1H, H-2), 3.85–3.66 (m, 4H, H6a, H6b, H-5, H-4); 13C NMR (100 MHz, CDCl3) δ 167.4, 154.5, 137.8, 133.7, 133.5, 133.1, 132.1, 131.9, 130.1, 129.1, 129.0, 128.8, 128.6, 128.3, 127.9, 127.6, 95.4, 86.9, 78.6, 74.3, 73.6, 70.4, 69.9, 55.0, 54.7; HRMS-ESI [M + Na]+ calcd for C29H28 Cl3NO7S 662.0544, found 662.0540.
Phenyl-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1 → 4)-3-O-benzoyl-6-O-benzyl-2-deoxy-2-(2,2,2-trichloroethoxycarbonylamino)-1-thio-β-D-glucopyranoside (16)
TMSOTf (25 μL, 0.01 mmol) was added dropwise to a suspension of imidate 15 (0.70 g, 1.42 mmol), acceptor 14 (0.91 g, 1.42 mmol) and 3 Å MS (0.3 g) at −50 °C and the reaction mixture was stirred at the same temperature for 3 h. Then the mixture was diluted with CH2Cl2 filtered through Celite and concentrated. The residue was purified by silica gel column chromatography (25% ethyl acetate:pet ether) to give the desired product 16 as a foam (0.65 g, 60%). Along with the product some amount of aglycone transferred product (phenyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-galactopyranoside) was also isolated (15%) which was confirmed by 1H and 13C NMR: [α]20D = −11.7 (c 2.3, CHCl3); IR (CHCl3) ν 2925, 1750, 1369, 1222, 1060, 760 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 7.3 Hz, 2H, ArH), 7.55–7.54 (m, 3H, ArH), 7.40–7.33 (m, 7H, ArH), 7.28–7.26 (m, 3H, ArH), 6.03 (d, J = 9.6 Hz, 1H, NH), 5.47 (t, J = 9.6 Hz, 1H, H-3), 5.09 (bs, 1H, H-4′), 4.97–4.93 (m, 2H, H-1, H-2′), 4.77–4.75 (m, 2H, H-3′), 4.67 (d, J = 12.0 Hz, 1H, CHHPh), 4.57 (d, J = 12.0 Hz, 1H, CHHPh), 4.51–4.45 (m, 3H, CH2 of NHTroc, H-1′), 4.13–4.03 (m, 2H, H-4, H-2), 3.82–3.73 (m, 2H, H-6a′ & H-6b′), 3.67–3.66 (m, 1H, H-5), 3.42–3.36 (m, 3H, H-6a & H-6b, H-5′), 1.97 (s, 6H, COCH3), 1.95 (s, 3H, COCH3), 1.92 (s, 3H, COCH3); 13C NMR (125 MHz, CDCl3) δ 170.2, 170.1, 168.9, 166.4, 154.4, 137.8, 133.4, 132.9, 131.7, 129.9, 129.6, 129.0, 128.6, 128.4, 128.2, 128.1, 127.5, 100.2, 95.4, 86.4, 78.8, 76.9, 74.7, 74.6, 74.3, 73.8, 70.9, 70.3, 69.3, 67.6, 66.5, 60.3, 54.9, 20.8, 20.7, 20.6, 20.3; HRMS-ESI [M + Na]+ calcd for C43H46Cl3NO16S 994.1476, found 994.1419.
N-(Benzyloxycarbonyl)-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1 → 4)-3-O-benzoyl-6-O-benzyl-2-deoxy-2-(2,2,2-trichloroethoxycarbonylamino)-β-D-glucopyranosyl-(1 → 3)-4-O-acetyl-2,6-di-O-benzoyl-β-D-galactopyranosyl-(1 → 3)-2-azido-2-deoxy-4-O-acetyl-6-O-t-butyldimethylsilyl-α-D-galactopyranosyl)-L-serine methylester (17)
To a suspension of donor 16 (0.13 g, 0.13 mmol), acceptor 11 (0.12 g, 0.10 mmol) and 3 Å MS (0.2 g) in CH2Cl2 (3 mL), NIS (0.09 g, 0.66 mmol) was added. After that TMSOTf (12 μL, 0.06 mmol) was added slowly dropwise at 0 °C and the reaction mixture was kept for stirring at rt for 2 h. After consumption of the starting material, Et3N (1.0 mL) was added to quench the reaction. Then the reaction mixture was filtered through Celite, washed with Na2S2O3, the separated organic layer was dried over Na2SO4 and concentrated. The crude product was purified by column chromatography on silica gel (40% ethyl acetate:pet ether) to afford 17 as a white foam (0.17 g, 80%): [α]20D = +25.1 (c 1.0, CHCl3); IR (CHCl3) ν 2930, 2110, 1749, 1369, 1228, 1070, 758, 711, 668, 504 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.07–8.05 (m, 4H, ArH), 7.94–7.93 (m, 2H, ArH), 7.64–7.30 (m, 29H, ArH), 5.64–5.61 (m, 2H, NH), 5.53–5.52 (m, 1H), 5.43 (dd, J = 9.6, 8.3 Hz, 1H), 5.19 (dd, J = 10.0, 9.8 Hz, 1H), 5.15–5.06 (m, 3H), 4.94 (dd, J = 10.1, 8.1 Hz, 1H), 4.84–4.82 (m, 2H), 4.78–4.74 (m, 2H), 4.70–4.68 (m, 1H), 4.58–4.54 (m, 3H), 4.50–4.42 (m, 3H), 4.39–4.36 (m, 1H), 4.07–4.00 (m, 5H), 3.99–3.91 (m, 3H), 3.75–3.72 (m, 2H), 3.71 (s, 3H, CO2CH3), 3.60–3.54 (m, 3H), 3.49–3.38 (m, 4H), 2.23 (s, 3H, COCH3), 1.98 (s, 3H, COCH3), 1.97 (s, 3H, COCH3), 1.92 (s, 6H, COCH3), 1.90 (s, 3H, COCH3), 1.03 (s, 9H, (CH3)3CSi); 13C NMR (125 MHz, CDCl3) δ 170.4, 170.3, 170.24, 170.2, 170.1, 169.4, 169.1, 166.1, 165.9, 164.9, 156.1, 154.0, 138.1, 136.1, 135.75, 135.7, 133.6, 133.5, 133.3, 133.2, 129.9, 129.8, 129.7, 129.5, 128.7, 128.6, 128.5, 128.47, 128.4, 128.0, 127.9, 127.8, 102.2, 101.8, 100.3, 98.9, 95.4, 75.0, 74.0, 73.9, 72.6, 71.5, 71.2, 71.0, 70.5, 69.6, 69.3, 69.1, 68.8, 67.6, 67.4, 66.6, 62.7, 62.3, 60.5, 59.5, 56.3, 54.3, 52.9, 26.8, 21.0, 20.83, 20.8, 20.7, 20.6, 20.5, 19.2; HRMS-ESI [M + Na]+ calcd for C95H104Cl3N5O34Si 2016.5297, found 2016.5254.
N-(Benzyloxycarbonyl)-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1 → 4)-3-O-benzoyl-6-O-benzyl-2-deoxy-2-(2,2,2-trichloroethoxycarbonylamino)-β-D-glucopyranosyl-(1 → 3)-4-O-acetyl-2,6-di-O-benzoyl-β-D-galactopyranosyl-(1 → 3)-2-azido-2-deoxy-4,6-di-O-acetyl-α-D-galactopyranosyl)-L-serine methylester (18)
A solution of TBAF (1.3 mL, 4.48 mmol, 1 M solution in THF) and AcOH (0.1 mL, 2.26 mmol) with pH 7 was added at 0 °C to a clear solution of 17 (0.12 g, 0.06 mmol) in THF (6.8 mL) and the reaction mixture was stirred at rt overnight. After complete consumption of starting material, solvents were removed in vacuo and the reaction mixture was azeotroped twice with toluene (2 × 5 mL). The crude product obtained after solvent evaporation was dissolved in pyridine (2.0 mL) and Ac2O (0.7 mL, 7.42 mmol) and kept for stirring. After consumption of the starting material, the solvent was concentrated in vacuo and chromatographed on silica gel (60% ethyl acetate:pet ether) to obtain 18 as a viscous liquid (0.09 g, 87%): [α]20D = +8.4 (c 0.1, CHCl3); IR (CHCl3) ν 3019, 2110, 1518, 1424, 1217, 759, 669, 627 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.10–8.01 (m, 4H, ArH), 7.99–7.92 (m, 2H, ArH), 7.61–7.27 (m, 19H, ArH), 5.73 (d, J = 8.1 Hz, 1H, NH), 5.63 (d, J = 3.1 Hz, 1H), 5.48–5.42 (m, 2H), 5.21–5.17 (m, 2H), 5.14–5.07 (m, 2H), 4.98–4.91 (m, 2H), 4.82–4.79 (m, 2H), 4.76–4.73 (m, 2H), 4.70–4.67 (m, 1H), 4.62–4.53 (m, 3H), 4.51–4.43 (m, 4H), 4.38–4.35 (m, 1H), 4.05–3.95 (m, 8H), 3.92–3.85 (m, 2H), 3.71 (s, 3H, CO2CH3), 3.56–3.53 (m, 2H), 3.47–3.36 (m, 4H), 2.21 (s, 3H, COCH3), 2.09 (s, 3H, COCH3), 2.00 (s, 3H, COCH3), 1.96 (s, 3H, COCH3), 1.90 (s, 6H, COCH3), 1.89 (s, 3H, COCH3); 13C NMR (125 MHz, CDCl3) δ 170.3, 170.2, 170.15, 170.13, 170.1, 169.7, 169.1, 166.2, 165.8, 164.8, 155.9, 154.0, 138.1, 136.1, 133.6, 133.5, 133.3, 129.83, 129.81, 129.7, 129.6, 129.5, 129.2, 128.7, 128.6, 128.57, 128.5, 128.44, 128.4, 128.3, 128.2, 128.1, 128.04, 128.0, 102.2, 102.0, 100.3, 99.0, 95.4, 75.0, 74.7, 74.3, 74.1, 74.0, 73.9, 72.5, 71.5, 71.0, 70.9, 70.5, 69.5, 69.4, 69.3, 69.1, 68.0, 67.6, 67.3, 66.5, 62.7, 62.5, 60.4, 59.1, 56.3, 54.4, 52.9, 20.2, 20.8, 20.7, 20.6, 20.5; HRMS-ESI [M + Na]+ calcd for C81H88Cl3N5O35 1820.4219, found 1820.4216.
N-(Benzyloxycarbonyl)-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1 → 4)-2-acetamido-2-deoxy-3-O-benzoyl-6-O-benzyl-β-D-glucopyranosyl-(1 → 3)-4-O-acetyl-2,6-di-O-benzoyl-β-D-galactopyranosyl-(1 → 3)-2-acetamido-2-deoxy-4,6-di-O-acetyl-α-D-galactopyranosyl)-L-serine methylester (19)
To a solution of compound 18 (0.06 g, 0.03 mmol) in EtOAc (3.4 mL) was added zinc (1.0 g, 16.04 mmol), acetic acid (550 μL, 9.65 mmol) and the solution was stirred at rt for 1 h. The reaction mixture was filtered through Celite and washed with EtOAc. To the filtrate was added Ac2O (605 μL) and the resulting mixture was stirred at rt for 15 h. The reaction mixture was concentrated and chromatographed on silica gel (90% ethyl acetate:pet ether) to give 19 as a colourless foam (0.04 g, 75%): [α]20D = +10.4 (c 0.1, CHCl3); IR (CHCl3) ν 3686, 3019, 1745, 1519, 1215, 1027, 928, 761, 669 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.06–8.01 (m, 4H, ArH), 7.93 (d, J = 7.3 Hz, 2H, ArH), 7.62–7.31 (m, 19H, ArH), 5.60–5.59 (m, 2H), 5.44–5.38 (m, 3H), 5.22–5.05 (m, 4H), 4.99–4.84 (m, 3H), 4.75–4.69 (m, 4H), 4.63 (d, J = 8.2 Hz, 1H), 4.45–4.31 (m, 6H), 4.07–3.74 (m, 10H), 3.71–3.68 (m, 2H), 3.65 (s, 3H, COOCH3), 3.57–3.30 (m, 4H), 2.17 (s, 3H, COCH3), 2.03 (s, 3H, COCH3), 2.00 (s, 3H, COCH3), 1.94 (s, 3H, COCH3), 1.90 (s, 3H, COCH3), 1.89 (s, 6H, COCH3), 1.42 (s, 3H, CH3), 1.35 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ 170.6, 170.5, 170.3, 170.26, 170.20, 170.16, 170.09, 169.2, 166.3, 166.1, 164.8, 155.9, 138.1, 136.0, 134.1, 133.5, 133.3, 129.9, 129.8, 129.6, 129.4, 129.2, 128.8, 128.7, 128.63, 128.6, 128.54, 128.5, 128.1, 128.03, 128.0, 102.0, 100.8, 100.3, 98.5, 71.8, 70.9, 70.5, 69.4, 69.3, 69.2, 68.4, 67.9, 67.8, 67.5, 66.6, 63.0, 62.6, 60.4, 54.6, 54.4, 52.7, 49.0, 22.7, 22.5, 20.9, 20.8, 20.7, 20.6, 20.5; HRMS-ESI [M + Na]+ calcd for C82H93N3O35 1702.5482, found 1702.5481.
N-(Benzyloxycarbonyl)-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1 → 4)-2-acetamido-2-deoxy-3-O-benzoyl-β-D-glucopyranosyl-(1 → 3)-4-O-acetyl-2,6-di-O-benzoyl-β-D-galactopyranosyl-(1 → 3)-2-acetamido-2-deoxy-4,6-di-O-acetyl-α-D-galactopyranosyl)-L-serine methylester (20)
A solution of NaBrO3 (0.04 g, 0.32 mmol) in water (1.2 mL) was added to a clear solution of 19 (0.09 g, 0.05 mmol) in EtOAc (3.0 mL). To this biphasic layer a solution of Na2S2O4 (0.05 g, 0.32 mmol) in water (1.8 mL) was added dropwise over 5 min. After 45 min, reaction mixture was quenched with aq. Na2S2O3 solution and extracted with EtOAc (3 × 30 mL). Combined organic layers dried over Na2SO4, concentrated and chromatographed on silica gel (5% methanol:ethyl acetate) to afford the desired product 20 as a foam (70 mg, 82%): [α]20D = −6.4 (c 0.1, CHCl3); IR (CHCl3) ν 3415, 2931, 1746, 1656, 1218, 1071, 917, 769, 713, 481 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.10–7.97 (m, 6H, ArH), 7.60–7.39 (m, 14H, ArH), 5.8 (bs, 1H), 5.60 (d, J = 7.7 Hz, 1H, NH), 5.46–5.35 (m, 4H), 5.16–5.03 (m, 5H), 4.89–4.86 (m, 3H), 4.75–4.74 (m, 1H), 4.61–4.56 (m, 2H), 4.49–4.34 (m, 2H), 4.20–4.02 (m, 5H), 3.96–3.72 (m, 8H), 3.67 (s, 3H, COOCH3), 3.64–3.59 (m, 1H), 3.47–3.36 (m, 3H), 2.25 (s, 3H, COCH3), 2.09 (s, 3H, COCH3), 2.08 (s, 3H, COCH3), 2.03 (s, 3H, COCH3), 1.98 (s, 3H, COCH3), 1.94 (s, 3H, COCH3), 1.94 (s, 3H, COCH3), 1.44 (s, 3H, CH3), 1.03 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ 171.1, 170.6, 170.3, 170.2, 170.1, 170.0, 169.7, 166.2, 165.9, 164.8, 155.9, 134.0, 133.7, 133.6, 133.5, 130.2, 129.9, 129.8, 129.7, 129.5, 129.4, 129.1, 128.8, 128.7, 128.6, 128.5, 101.8, 101.2, 100.9, 98.5, 71.3, 71.0, 70.6, 69.6, 69.4, 69.3, 68.3, 67.9, 67.5, 66.5, 63.0, 61.5, 60.1, 55.3, 54.4, 52.8, 49.0, 22.5, 22.3, 21.3, 20.9, 20.8, 20.73, 20.7, 20.6; HRMS-ESI [M + Na]+ calcd for C75H87N3O35 1612.5007, found 1612.5012.
N-(Benzyloxycarbonyl)-O-(β-D-galactopyranosyl-(1 → 4)-2-acetamido-2-deoxy-6-sulfo-β-D-glucopyranosyl-(1 → 3)-β-D-galactopyranosyl-(1 → 3)-2-acetamide-2-deoxy-α-D-galactopyranosyl)-L-serine (1)
To a solution of 20 (0.03 mg, 0.02 mmol) in dry pyridine (1 mL) was added SO3·NEt3 (40 mg, 0.22 mmol). The reaction was stirred for 48 h at rt. After complete consumption of the starting material, the reaction mixture was concentrated and co-evaporated with toluene (3 × 5 mL). A solution of NaOMe (24 mg) in MeOH (1 mL) was added to the crude product in MeOH (1 mL) and water (1 mL) at 55 °C (pH 10). After stirring at the same temperature for 30 h, the reaction mixture was neutralized by adding AcOH until the pH adjusted to 6. The reaction mixture was then concentrated and purified by silica gel chromatography (7:2:1 ethyl acetate:MeOH:H2O) to give 1 as a waxy solid (17 mg, 80%): 1H NMR (400 MHz, CDCl3) δ 7.33–7.30 (m, 5H, ArH), 4.60–4.58 (m, 2H), 4.38 (d, J = 7.8 Hz, 1H), 4.32 (d, J = 7.8 Hz, 1H), 4.28–4.25 (m, 1H), 4.20–4.16 (m, 2H), 4.10–4.01 (m, 3H), 3.91 (d, J = 3.0 Hz, 1H), 3.90–3.89 (m, 1H), 3.86–3.79 (m, 3H), 3.76–3.61 (m, 5H), 3.60–3.44 (m, 13H), 3.43–3.38 (m, 3H), 1.89 (s, 3H, CH3), 1.79 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ 182.5, 181.5, 174.9, 174.6, 157.7, 136.5, 128.8, 128.7, 128.4, 127.8, 127.7, 104.7, 102.7, 98.0, 82.3, 75.4, 74.6, 72.6, 72.5, 72.2, 71.0, 70.8, 69.7, 68.8, 68.7, 68.6, 68.4, 67.1, 66.6, 61.1, 55.2, 48.5, 23.3, 22.3, 22.2, 20.1; HRMS-ESI [M + Na]+ calcd for C39H58N3NaO28S 1094.2717, found 1094.2714.
Acknowledgements
We thank Department of Science and Technology (Grant no. SR/S1/OC-40/2009), Council of Scientific and Industrial Research (Grant no. 01(2376)/10/EMR-II) and Board of Research in Nuclear Sciences (Grant no. 2013/37C/51/BRNS) for financial support. A. B. and M. E. thank UGC and CSIR-New Delhi for their fellowships.Notes and references
- T. Ju, V. I. Otto and R. D. Cummings, Angew. Chem., Int. Ed., 2011, 50, 1770–1791 CrossRef CAS PubMed.
- H. C. Hang and C. R. Bertozzi, Bioorg. Med. Chem., 2005, 13, 5021–5034 CrossRef CAS PubMed.
- A. Varki, R. Kannagi and B. P. Toole, Glycosylation Changes in Cancer, in Essentials of Glycobiology, ed. A. Varki, Cold Spring Harbor Laboratory Press, New York, 2nd edn, 2009 Search PubMed.
- S. J. Danishefsky and J. R. Allen, Angew. Chem., Int. Ed., 2000, 39, 836–863 CrossRef CAS.
- C. Brocke and H. Kunz, Bioorg. Med. Chem., 2002, 10, 3085–3112 CrossRef CAS.
- T. Buskas, S. Ingale and G.-J. Boons, Angew. Chem., Int. Ed., 2005, 44, 5985–5988 CrossRef CAS PubMed.
- A. Kaiser, N. Gaidzik, T. Becker, C. Menge, K. Groh, H. Cai, Y.-M. Li, B. Gerlitzki, E. Schmitt and H. Kunz, Angew. Chem., Int. Ed., 2010, 49, 3688–3692 CrossRef CAS PubMed.
- Z. Yin and X. J. Huang, J. Carbohydr. Chem., 2012, 31, 143–186 CrossRef CAS PubMed.
- U. Westerlind, A. Hobel, N. Gaidzik, E. Schmitt and H. Kunz, Angew. Chem., Int. Ed., 2008, 47, 7551–7556 CrossRef CAS PubMed.
- J. Yeh, N. Hiraoka, B. Petryniak, J. Nakayama, L. G. Ellies, D. Rabuka, O. Hindsgaul, J. D. Marth, J. B. Lowe and M. Fukuda, Cell, 2001, 105, 957–969 CrossRef CAS.
- P. R. Streeter, B. T. Rouse and E. C. Butcher, J. Cell Biol., 1988, 107, 1853–1862 CrossRef CAS.
- S. Hemmerich, E. C. Butcher and S. D. Rosen, J. Exp. Med., 1994, 180, 2219–2226 CrossRef CAS.
- F. Bélot, D. Rabuka, M. Fukuda and O. Hindsgaul, Tetrahedron Lett., 2002, 43, 7743–7747 CrossRef.
- L. A. Marcaurelle, M. R. Pratt and C. R. Bertozzi, ChemBioChem, 2003, 4, 224–228 CrossRef CAS PubMed.
- M. R. Pratt and C. R. Bertozzi, Org. Lett., 2004, 6, 2345–2348 CrossRef CAS PubMed.
- M. Emmadi and S. S. Kulkarni, J. Org. Chem., 2011, 76, 4703–4709 CrossRef CAS PubMed.
- M. Emmadi and S. S. Kulkarni, Org. Biomol. Chem., 2013, 11, 4825–4830 CAS.
- M. Emmadi and S. S. Kulkarni, Nat. Protoc., 2013, 8, 1870–1889 CrossRef CAS PubMed.
- K.-L. Chang, M. M. L. Zulueta, X. A. Lu, Y.-Q. Zhong and S.-C. Hung, J. Org. Chem., 2010, 75, 7424–7427 CrossRef CAS PubMed.
- Y. Cai, C.-C. Ling and D. R. Bundle, J. Org. Chem., 2009, 74, 580–589 CrossRef CAS PubMed.
- C. H. Hassall and J. O. Thomas, J. Chem. Soc. C, 1968, 1495–1501 RSC.
- R. M. van Well, K. P. R. Kartha and R. A. Field, J. Carbohydr. Chem., 2005, 24, 463–474 CrossRef CAS PubMed.
- G. Blatter and J. C. Jacquinet, J. Carbohydr. Chem., 1996, 288, 109–125 CAS.
- R. V. Stick and K. A. Stubbs, Tetrahedron: Asymmetry, 2005, 16, 321–335 CrossRef CAS PubMed.
- S. Nakashima, H. Ando, A. Imamura, N. Yuki, H. Ishida and M. Kiso, Chem.–Eur. J., 2011, 17, 588–597 CrossRef CAS PubMed.
- M. P. DeNinno, J. B. Etienne and K. C. Duplantier, Tetrahedron Lett., 1995, 36, 669–672 CrossRef CAS.
- R. R. Schmidt and M. Stumpp, Liebigs Ann. Chem., 1983, 1249–1256 CrossRef CAS.
- H. V. Ravi Kumar, K. Naruchi, R. Miyoshi, H. Hinou and S.-I. Nishimura, Org. Lett., 2013, 15, 6278–6281 CrossRef CAS PubMed.
- M. Adinolfi, L. Guariniello, A. Iadonisi and L. Mangoni, Synlett, 2000, 1277–1278 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C and 2D NMR spectra. See DOI: 10.1039/c4ra12631a |
| This journal is © The Royal Society of Chemistry 2014 |