
Real-time detection of telomerase activity in cancer cells using a label-free electrochemical impedimetric biosensing microchip†
Lisandro Cunci*ab,
Marina Martinez Vargasac,
Roman Cuncid,
Ramon Gomez-Morenoac,
Ivan Perezab,
Abel Baerga-Ortizac,
Carlos I. Gonzalezace and
Carlos R. Cabrera*ab
aMolecular Sciences Research Building, University of Puerto Rico, 1390 Ponce de Leon Ave., STE. 2, San Juan, Puerto Rico 00926-2614, USA. E-mail: lisandro.cunci@upr.edu; carlos.cabrera2@upr.edu; Fax: +1-787-522-2150; Tel: +1-787-523-5319
bDepartment of Chemistry and Center for Advanced Nanoscale Materials, University of Puerto Rico, Río Piedras Campus, P.O Box 23346, San Juan, Puerto Rico 00931-3346, USA
cDepartment of Biochemistry, University of Puerto Rico, Medical Sciences Campus, P.O. Box 365067, San Juan, Puerto Rico 00936-5067, USA
dDepartment of Computer Engineering, Buenos Aires Institute of Technology, Av. Eduardo Madero 399, C1106ACD, Buenos Aires, Argentina
eDepartment of Biology, University of Puerto Rico, Río Piedras Campus, P.O. Box 23360, San Juan, Puerto Rico 00931-3360, USA
First published on 15th October 2014
Abstract
The enzyme telomerase is present in about 85% of human cancers which makes it not only a good target for cancer treatment but also an excellent marker for cancer detection. Using a single stranded DNA probe specific for telomerase binding and reverse transcription tethered to an interdigital gold electrode array surface, the chromosome protection provided by the telomerase was replicated and followed by electrochemical impedance spectroscopy as an unlabeled biosensor. Using this system designed in-house, easy and affordable, impedance measurements were taken while incubating at 37 °C and promoting the probe elongation. This resulted in up to 14-fold increase in the charge transfer resistance when testing a telomerase-positive nuclear extract from Jurkat cells compared to the heat-inactivated telomerase-negative nuclear extract. The electron transfer process at the Au electrodes was studied before the elongation, at different times after the elongation, and after desorption of non-specific binding.
Introduction
Human chromosomes have at their very end DNA sequences known as telomeres, which are shortened in every cell replication cycle. These telomeres consist of tandem repeats of species-dependent G-rich sequences (particularly in vertebrates, 5′-TTAGGG-3′). Due to the continuous shortening of these DNA sequences during cell replication, “healthy” somatic cells cannot go beyond the Hayflick limit of cell division.1 However, the majority of cancer cells (approximately 85%) have found a loophole, activating the telomerase enzyme whose composition was discovered in 2007 by S. B. Cohen et al.2 Cancer cells that do not activate telomerase, use an alternative mechanism known as alternative lengthening of telomeres (ALT), which is still not completely understood.3 The vast presence and activity of the enzyme telomerase in cancer cells makes it a suitable target for diagnosis and treatment. Also, inhibition of Telomerase activity is known to induce tumor cell growth arrest. In immortal cells, such as the Jurkat T cell line used in this study, Telomerase aids in the maintenance of telomeres.Cancer cell biosensors have been one of the main topics in the scientific literature during the last few years.4–8 Their potential use at the point of care can translate into an early onset of treatment that may change the prognosis of the patients. Several biosensors for the detection of telomerase as a cancer marker have been reported within the last few years,6,7,9–12 and two recent reviews.13,14 The most promising biosensor platforms that specifically detect telomerase rely on the principle of the elongation of single stranded DNA probes with specific sequences,15 such as 5′-TTAGGG-3′ for vertebrates. Using this principle, many electrochemical telomerase biosensors have been reported using differential pulse voltammetry,16–19 electroluminescence,20,21 and chronocoulometry,22,23 with only one that uses electrochemical impedance spectroscopy (EIS) for a dual purpose: for surface characterization and as the biosensor signal transducer.24
There are numerous advantages of using EIS in biosensor design, such as high sensitivity25,26 accuracy, rapid detection, compatibility with microfabrication technology27 and real-time measurements.28 In the detection of telomerase, EIS has been used in one instance24 where a ssDNA oligomer probe was tethered to the surface of a gold disk electrode, followed by incubation of the electrode in a HeLa cell extract for several hours. Then, the electron transfer reaction of the ferricyanide/ferrocyanide redox couple was measured in a different buffer by EIS, and the electron transfer resistance was correlated to the elongation of the probe. However, these reported biosensor configurations only considered the ferricyanide/ferrocyanide redox reaction and none have taken advantage of the vast diversity of molecules already in total/nuclear cell extracts and buffer solutions.
In this work, we designed easy software and constructed hardware for an in situ incubation while detecting the presence of the telomerase enzyme as a cancer marker by taking advantage of its reverse transcriptase activity measured in real time. For these experiments, we used Jurkat T cells which have been useful in studies of Telomerase activity regulation and in understanding the mechanisms of differential susceptibility of several cancers to radiation and other treatments.29–31 A nuclear extract from Jurkat cells in the absence of an additional redox couple was used as the telomerase-positive sample, and a solution heat-treated at 95 °C to inactivate the telomerase enzyme as the control. This work focused on the development of a proof-of-concept, straightforward detection of telomeric activity in under 20 minutes with a robust, powerful and scalable method, using an interdigital biosensor microchip and avoiding the need of a reference electrode. We propose this system as a step towards inexpensive and faster cancer detection at the point of care.
Experimental
Software and hardware for temperature control
The software was made using all open source components, which are available in the internet free of cost. The programming language for the desktop client is Java Version 7, which can be downloaded from Oracle without charge. The serial protocol used for communication with the circuit and the main algorithm developed for controlling it are novel. The rest of the functionality was created using pre-existing libraries. The graphs were plotted using JFreeChart and the low-level serial communication is performed using NeuronRobotics's nrjavaserial library. All the libraries used to make the software have business-friendly licenses, and do not impose any further restriction. An image of the software is shown in the ESI, Fig. SI1.†The hardware was designed to be easy to implement and inexpensive. Our temperature controller was built with commercially available components in a way that would allow any user to make their own with off-the-shelf components. A scheme of the circuit is presented in Fig. SI2,† and most elements may be obtained as samples for testing from each manufacturer. The circuit was soldered by hand and an explanation of how it functions is provided in the ESI.† We avoided the use of Labview and costly equipments for something as simple as temperature control for in situ incubation. On the other hand, we used a potentiostat for accurate EIS measurements but simple impedance measurement circuits can be modified to work with this system while reporting to the same software.
Microchip fabrication
The biosensor interdigital gold electrode array microchip was fabricated by us at the Conte Nanotechnology Cleanroom Laboratory that is part of the Center of Hierarchical Manufacturing at the University of Massachusetts, Amherst. We used 500 μm thick, 100 mm dia., single side polished, 〈100〉 oriented silicon wafers where we deposited ca. 200 nm of SiO2 by Plasma-Enhanced Chemical Vapor Deposition as isolation. Then, we used S1813 (Shipley) photoresist which was spinned at 3000 rpm for 60 seconds before pre-baking for 60 seconds at 115 °C. A Suss Microtec MA6 Mask Aligner with a 400 nm, 350 W, UV lamp was used with our photolithography mask (Front Range Photomask). Then, we deposited 5 nm of Ti and 150 nm of Au in a CHA SE-600 electron beam evaporator, after which we proceeded with the photoresist lift-off in acetone.Positive and control nuclear extracts preparation
DNA self-assembled monolayer deposition
The immobilization of the ssDNA probe was carried out as follows. After cleaning in piranha solution for 30 minutes, the interdigital electrode arrays (IDA) were exposed to an aqueous solution of 2 mM thioglycolic acid overnight, which formed a SAM that left an exposed carboxyl group. This group was reacted with a solution of EDC 20 mM and sulfo-NHS 50 mM for 2 hours after which a solution of 1 μM NH2-(CH2)6-TS (5′-NH2(CH2)6TTTTTTTTTTAATCCGTCGAGCAGAGTT-3′) (Integrated DNA Technologies) was deposited on the modified IDA and left overnight. Then, the IDA were washed carefully and treated with nanopure water, followed by drying with N2. After that, we carefully washed with 40 mM NH2OH·HCl and 0.05% SDS, and finally dried with N2 again to be used the same day. The O-ring rubbers were maintained in ethanol until needed for the electrochemical cell ensemble, when they were washed with nanopure water, dried with N2 and placed on the IDA. Finally, 20 μL of nuclear extract solution was added inside the O-ring and closed with a piece of clean glass slide. Fig. 1a shows a schematic of the procedure used for the tethering of TS to the interdigital gold surface electrodes. | ||
| Fig. 1 (a) Procedure for the tethering of the TS probes and (b) the mechanism responsible for the expected change in the impedance during incubation at 37 °C. | ||
Electrochemical measurements
The electrochemical experiments were done in an Autolab PGSTAT30 potentiostat. EIS experiments were conducted from 1.0 MHz to 0.1 Hz taking 40 measurements in logarithmic scale, with an amplitude of 10 mVp–p, single sine method, at 0.00 V applied voltage between the two electrodes. The open circuit voltages were taken just before the experiments from the potentiostat front panel, and they all measured equal or less than 0.02 V which was used to verify that both electrodes were similar before starting.DNA visualization
A solution of 10× TAE buffer (pH 8.18–8.29) was prepared by mixing 48.4 g of Tris-base (Sigma-Aldrich), 10.9 g of glacial acetic acid (Sigma-Aldrich), and 2.92 g of EDTA (Sigma-Aldrich) in 1 L of nanopure water. The dilution of 1 g of agarose (BioRad) was done in 100 mL of 1× TAE buffer to obtain a 1% agarose matrix. The agarose gel was stained by adding 10 μL of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× GelRed (Biotium) to visualize DNA under UV light at a wavelength of 302 nm. The agarose gel was placed in the electrophoresis chamber (BioRad) followed by adding 1× TAE buffer until the gel was completely submerged. DNA samples were prepared by mixing 15 μL of DNA with 1.5 μL of 5× nucleic acid sample loading buffer (BioRad). Each DNA sample was placed in its appropriate well, and run at 100 V (constant) for 40 minutes.
000× GelRed (Biotium) to visualize DNA under UV light at a wavelength of 302 nm. The agarose gel was placed in the electrophoresis chamber (BioRad) followed by adding 1× TAE buffer until the gel was completely submerged. DNA samples were prepared by mixing 15 μL of DNA with 1.5 μL of 5× nucleic acid sample loading buffer (BioRad). Each DNA sample was placed in its appropriate well, and run at 100 V (constant) for 40 minutes.
Results and discussion
The real-time biosensing of telomerase activity at an IDA microchip can be divided into three main parts: temperature control, biosensor microchip and electrochemical measurements. Fig. 1b shows the mechanism responsible for the expected change in the impedance during incubation at 37 °C. First, (1) the SAM of the TS probe specific for telomerase binding was formed at the interdigital electrodes surface, and it was in contact with the telomerase-positive nuclear extract solution at the moment of the addition. Then, (2) while the biosensor IDA microchip is incubating at 37 °C, the telomerase enzyme binds to the TS probes tethered to the gold surfaces due to its specificity and starts the TS elongation. These probes act as a specific substrate for telomerase due to the oligomer sequence 5′-TTAGGG-3′ recognized by its RNA component as a template used in the reverse transcriptase reaction. During incubation, additional (3) telomerase enzymes start binding to their substrate, TS, at the IDA surfaces and start the elongation, adding to the previously bound enzymes. The binding and growth exerted by the telomerase enzymes produce a blocking layer that hinders electrochemical surface reactions at the IDA, which was used as the signal change in the unlabeled biosensor microchip. Finally, (4) the telomerase enzymes continue elongating the probes. The blocking layer reaches a thickness where the kinetics of the electrochemical surface reactions is too slow. Any further change is too small to be measurable and the change in electron transfer reaches an asymptote.Temperature control
Our team designed an interdigital microelectrode Au array biosensor operated by an in-house custom made circuit and software which controls and records the temperature vs. time in situ. One important characteristic for the ssDNA probe elongation by the enzyme telomerase is the in situ incubation. Telomeres are elongated in the human body by this enzyme at ca. 37 °C which is the temperature selected for the biosensor microchip used in this study. Telomerase biosensors previously reported in the literature18,24 were placed inside an incubator or thermal cycler, depending on the experimental conditions assayed. Either, promoting the lengthening of a probe on a surface or the Telomeric Repeat Amplification Protocol (TRAP) assay,34 this later step was unavoidable before measurement and, in turn, for telomerase activity detection. Hence, the experiment needed to be stopped until the incubation period was completed, and tested either electrochemically or optically, among others. This resulted not only in the need of heavy equipment but also in the impossibility of real-time measurements while the oligonucleotide elongation is happening. Moreover, although procedures such as real time PCR may be used, they also need sophisticated and expensive equipment, labels and extensive purification procedures due to its reliance on fluorescence for measurement. Our design allows us to avoid these complications and to maintain temperature control measurements, and electrochemical detection of telomerase activity in real-time. Our effort to provide an inexpensive way to build an in-house custom made temperature controller was not only to allow the scientific community to test a real-time biosensing microchip, but also to provide flexibility and avoid the usual boundaries of commercial software packages.IDA electrode and supporting system
Our group designed a biosensor microchip with an integrated resistance temperature detector (RTD), which consists of a 10 μm path in a serpentine-shaped pattern that is accessed through the internal contacts of the USB-type connector. In addition, the biosensor microchip consisted of two 100-finger interdigital array (IDA) electrodes connected to the external contacts. These elements were connected using a standard type-A female USB cable. A finished biosensor microchip is shown in Fig. 2a, with a total size of 1 cm × 2 cm × 500 μm. The resistance measured at the two middle gold connectors is previously calibrated and correlates with the temperature of the IDA biosensor. The RTD was designed to be on the same substrate next to the sensor array for accurate temperature control. | ||
| Fig. 2 Images of (a) the biosensor microchip made by photolithography, (b) and (c) the system used for heating and connecting the biosensor, and (d) a scheme of our device closed and ready for measurements. Scanning electron microscopy images of the interdigital gold electrodes array at (e) 30× and (f) 550×. | ||
The accuracy in temperature measurement during detection in real time is very critical. Thermocouples, thermistors and other commercially available temperature sensors are covered with a packaging that delays precise temperature measurements resulting in inaccurate values, and have different thermal properties, which may result in unknown steady-state temperatures. Moreover, the delay due to the volumetric heat capacity can prevent rapid changes in temperature required for accurate control.
Each of the two electrodes of the interdigital microarray connected to the external pins are used for impedance measurement. The impedance was measured in a two electrode configuration, with the working electrode connected on the left electrode and the counter and reference electrodes together connected on the right electrode.
A support system was built for the microchip shown in Fig. 2 (Panels b, c, and d). Fig. 2 (Panels b and c) show the two aluminum sheets placed firmly together with screws and nuts, with a 1′′ dia. silicone flexible heater used for rapid changes in temperature (All Flex, Inc.) in contact with the biosensor microchip, which provides a fast temperature response. The reaction chamber was built using a rubber O-ring that enclosed the microelectrode sensor array with a glass slide on top. The volumetric capacity depends on the size of the O-ring, commonly being close to 20 μL. Even though 37 °C does not promote fast evaporation, since we used a few microliters, we found out that the reaction chamber was required in order to prevent solution evaporation during the experiments and further change in concentration. A clear scheme of the setup is shown in Fig. 2d.
The design of the IDA microchip electrode maximizes the interaction between the electrodes.35,36 An IDA was designed with a finger width of 10 μm and a separation of 5 μm between the fingers. After construction, the finger width was ca. 10.5 μm and the separation between the fingers ca. 4.5 μm. A scanning electron microscope (SEM) image of the IDA is shown in Fig. 2e and a close-up image in Fig. 2f. The theoretical area of each electrode was designed to be ca. 4.5 mm2, and the perimeter of closest contact at 52 cm.
Electrochemical experiments
We designed an unlabeled biosensor microarray avoiding the addition of labeling redox coupled molecules to provide electron transfer reactions such as ferrocyanide/ferricyanide. Instead, we took advantage of human cancer cells and buffers to prepare an in vitro system that would allow for rapid unlabeled detection of telomerase activity in nuclear extracts.First, the IDA was treated with piranha solution to clean all organic materials left from the photolithography process. Then, we used CV at 100 mV s−1, from 0.3 V to 1.5 V vs. Ag|AgCl in 0.5 M H2SO4 to verify that the gold surface was cleaned and both electrodes were homogenous in surface area. The TS probe was tethered to the gold surface as explained above, and the electrodes were used the same day that were modified. The sensors were maintained at 37 °C with our temperature controller and a measurement was taken every 5 minutes with the potentiostat. The open circuit potentials between the two electrodes before starting the experiments were less than or equal to 0.02 V. The impedance measurements were conducted from 1 MHz to 0.1 Hz, with a voltage amplitude of 10 mV at a fixed voltage of 0.00 V between the two electrodes. Between measurements, the electrodes were left at open circuit to avoid any electron transfer.
Nyquist plots were used to compare the EIS measurements of the biosensor IDA microchip with the telomerase-positive and telomerase-negative nuclear extracts as shown in Fig. 3a and b, respectively. Both electrodes showed Voigt circuits with at least three reactions obtained from three peaks seen in the phase curve in the Bode plots (data not shown). This may be due to at least three different reactions, or more than one reaction occurring at both electrodes with different chemical kinetics. These reactions are provided by the vast compounds between the buffer C used for the nuclear extraction, and the nuclear extract itself, without the need of an external labeling agent. These results are consistent with a two electrode system where both electrodes are similar (although never equal) and differ, even slightly, from each other in their surface properties. In these Nyquist plots, only the reaction at the lowest frequencies is appreciated, which is the most affected by telomerase binding and elongation, and has the slowest kinetics. Analyzing this reaction, it is clear from these plots that both electrodes experience significant changes in impedance in less than 20 minutes. Fig. 3a and b showed half circles expanding with time, and means that the charge transfer resistance (Rct) is increased. Rct is correlated to the diameter of the half circle, and it is easily seen in these graphs that the increment in the diameter is larger for the telomerase-positive solution compared to the telomerase-negative solution. This increase can be analyzed quantitatively by modeling the data and plotting the change in percentage with regard to the initial measurement.
 | ||
| Fig. 3 Nyquist plots of the electrochemical impedance spectroscopy results for (a) the telomerase-negative solution and (b) telomerase-positive solution in TS modified interdigital Au electrodes while incubating at 37 °C, and (c) the comparison of their charge transfer resistance, Rct, with time. (d) Nyquist plot of an addition experiment with (e) the measurement of its Rct through time. | ||
Fig. 3c shows Rct calculated with the FRA Software (Autolab) versus time. Modeled with an asymptotic exponential equation (black line), the change in Rct is significant in only 20 minutes. The telomerase-positive as well as the telomerase-positive samples showed changes in Rct with time, however, these changes are significantly different when analyzed as a change in percentage compared to the initial measurement. The change in Rct in the telomerase-negative sample may be attributed to different reasons such as chemical changes in the solution and surface that occurred at 37 °C as well as temperature stabilization, that are also occurring at the telomerase-positive electrodes. For example, even though the O-ring provides a very good seal we may not prevent complete evaporation, which in turn changes the electrolyte concentration with time. Not only evaporation, but also, non-specific adsorptions can impact greatly the change in impedance. Since both electrodes experienced similar changes, we can attribute the difference in Rct vs. time to the elongation of the TS primer tethered to the IDA electrode surface.
These two electrodes differ in their absolute values due to variability in the construction and modification process. To verify these results, we repeated these experiments using a similar electrode but performing an additive experiment. We tested 10 μL of the heated control solution in our biosensor IDA microchip, and after 20 minutes, we added 10 μL of Jurkat nuclear extract solution with a total protein concentration of 763 μg mL−1. Fig. 3d and e show the Nyquist plot and the percent change in Rct with time, respectively. Again, the results follow a Voigt circuit with phase peaks similar to the electrodes discussed previously, and the diameter of the half circle increases with time, due to the elongation of the surface tethered primer TS. The percent change in Rct stays below 10% until 20 minutes where the telomerase-positive solution was added, and it raises up to ca. 45% in 15 minutes.
The tethering and elongation of the TS probe due to telomerase binding and reverse transcription with the reversibility of the electron transfer process at the Au surface was studied by cyclic voltammetry (CV). The CV was conducted in a 3 electrode system with both electrodes of the biosensor microchip used as the working electrode, a Pt wire as counter and Ag|AgCl as reference electrodes. A solution of 0.1 M PBS with 0.1 M KCl at pH 7.0, 1 mM of K3Fe(CN)6 and 1 mM K4Fe(CN)6 was used for the CV studies. Fig. 4 (Panels a, b and c) show the CV for Au, Au–TS, and Au–TS-elongated for 20 minutes at 37 °C with the telomerase-positive solution, respectively, from 10 to 300 mV s−1 in scan rates. Fig. 4d shows the CV for Au–TS-elongated after 4 months at room temperature in contact with telomerase-positive solution in a humidity saturated enclosure to avoid evaporation. Fig. 4e shows the CV for Au–TS-elongated (long time) for 4 months and stripped using 20 μL of 0.1 M PBS solution and chronoamperometry for 2 min at 1.3 V.37 This voltage was applied using one of the two IDA electrodes as working electrode, and connecting the second IDA electrode to the reference and counter electrode cables together. Prior to the experiments, these electrodes were washed thoroughly with nanopure water and 0.1 M PBS solution. These CV differentiate clearly in the shape of the oxidation and reduction peaks while after 4 months at room temperature the peaks are not detected for several scan rates. From these CV, the separation of the oxidation and reduction peaks (ΔEp–p), and the oxidation and reduction peak currents, iox and ired, respectively, were plotted versus the square root of the scan rates (v1/2) in Fig. 4 (Panels from f, g and h).
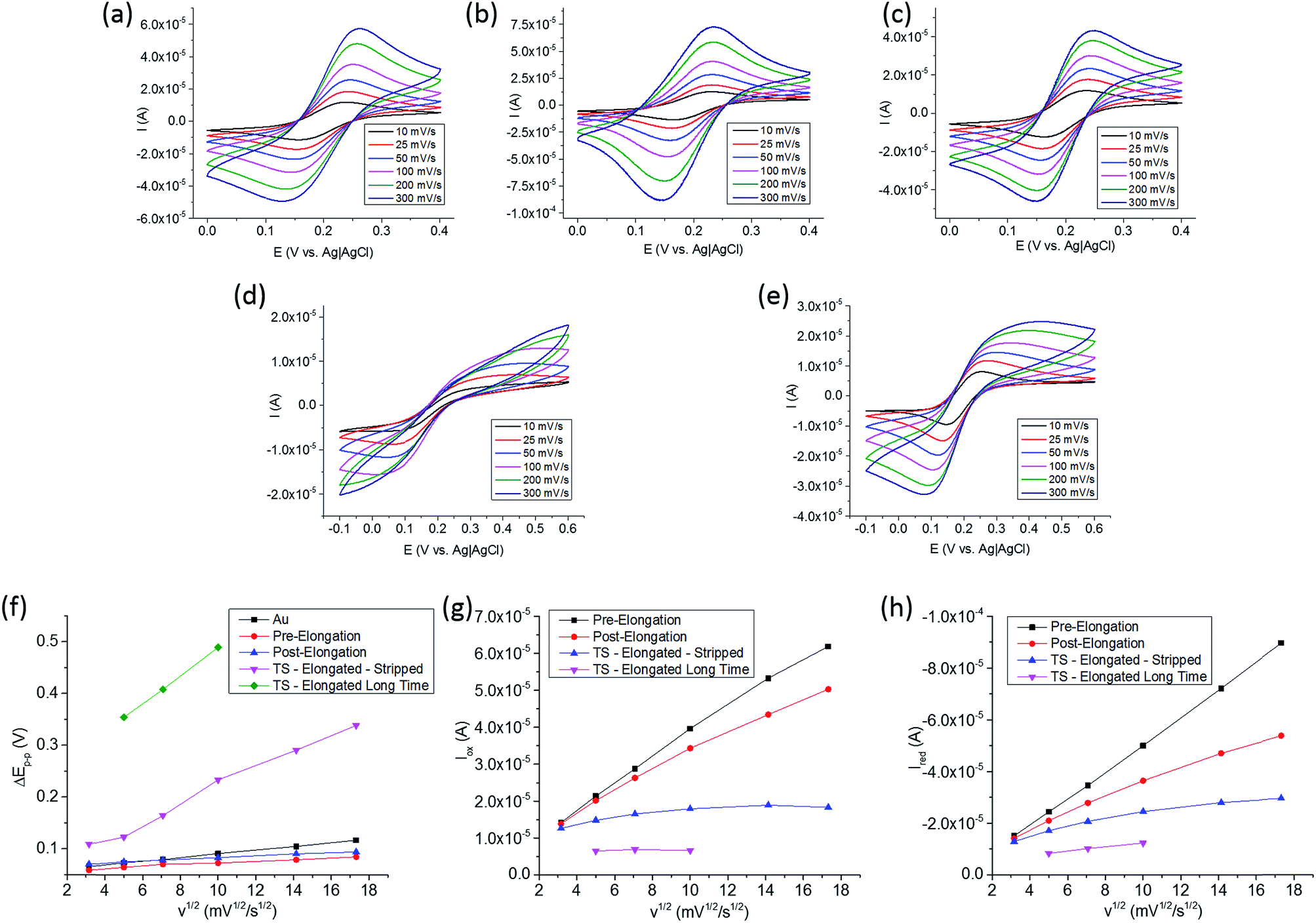 | ||
| Fig. 4 Cyclic voltammetry analysis of the interdigital electrodes array in 0.1 M PBS with 0.1 M KCl, 1.0 mM of K3Fe(CN)6 and 1.0 mM K4Fe(CN)6 at pH 7.0: (a) bare Au, (b) Au–TS before elongation, (c) Au–TS after elongation, (d) Au–TS after 4 months in contact with telomerase positive nuclear extract, and (e) “d” after 2 min at 1.3 V between interdigital electrodes for non-specific binding species desorption. Analysis of cyclic voltammetric data of (a) to (e) interdigital electrodes: (f) potential separation between anodic and cathodic peak currents, ΔEp–p, (g) anodic peak current, iox, and (h) cathodic peak current, ired versus v1/2. | ||
First, ΔEp–p decreases after the formation of the SAM compared to the clean gold electrode that is attributed to a more reversible electron transfer process due to faster kinetics. It is known that ssDNA has isoelectric point of 4.0–4.5 (ref. 38) with a negative charge when exposed to a pH 7.0 solution, and ferricyanide and ferrocyanide have negative charge which may suggest a slower electron transfer. However, we observed faster kinetics and enhanced reversibility as seen by L. Qingwen et al.39 when using a SAM of L-cysteine at Au and I. Feliciano-Ramos et al.40 when having a SAM of L-cysteine at Pd electrodes. Both groups worked at pH 7.0 and, although the carboxyl group has a negative charge, they attribute the enhanced reversibility to the local positive charge density of the amine. Therefore, even though TS has a net negative charge density, the localized positive charge densities of the amine groups may help the electron transfer process when the ssDNA is short enough, improving the reversibility as observed in our data. However, the reversibility of the electron transfer process in TS post-elongation, Fig. 4f, was affected by the increase of the ΔEp–p after 20 minutes in contact with the telomerase-positive solution. Compared to TS pre-elongation, the increment from 59 mV at 10 mV s−1, and 85 mV at 300 mV s−1, to 70 mV and 94 mV, respectively, is due to two mechanisms responsible for biosensing: (1) the telomerase binding to the TS probe and (2) the elongation of the TS probe at the electrode surface. In addition, the iox and ired peak currents were affected not only decreasing but also deviating from the linearity versus v1/2 as observed in the TS pre-elongation biosensor. This deviation from linearity at higher scan rates can be associated to a slower electron transfer process due to the blocking effect provided by the telomerase binding to the TS probe and its elongation.
After 4 months at room temperature, ΔEp–p increased dramatically not only due to the elongation but also probably due to non-specific surface interactions produced by the degradation products of the nuclear extract components. We applied a voltage of 1.3 V between the two electrodes during 2 min to desorb these species, and then, the connectors were swapped and the electrode used as working electrode was used as reference/counter electrode and vice versa, and 1.3 V was applied again for 2 min.37 The 20 μL PBS solution from the first and the second chronoamperometry, and a solution of the TS probe were used in an agarose gel electrophoresis (AGE) separation study. The results are shown in Fig. 5 next to the 10 kb DNA ladder. In the first column, we observed degradation fragments which were adsorbed to the electrode surface before the stripping procedures. In the second column, the absence of a line confirms that at least the majority of the adsorbed species were stripped from the electrode. In the third column, we observed one line at the bottom corresponding to the TS probe. The biosensor microchip after the stripping process should have a surface covered mostly by the elongated TS probe, as nothing else was observed in the second column of the AGE. After the chronoamperometry, the electron transfer process was faster with better reversibility as depicted in Fig. 4f (pink), shown by a decrease on the ΔEp–p, an increase in iox and ired, and a iox/ired peak current ratio closer to 1, as shown in Fig. 4 (Panels g and h, blue).
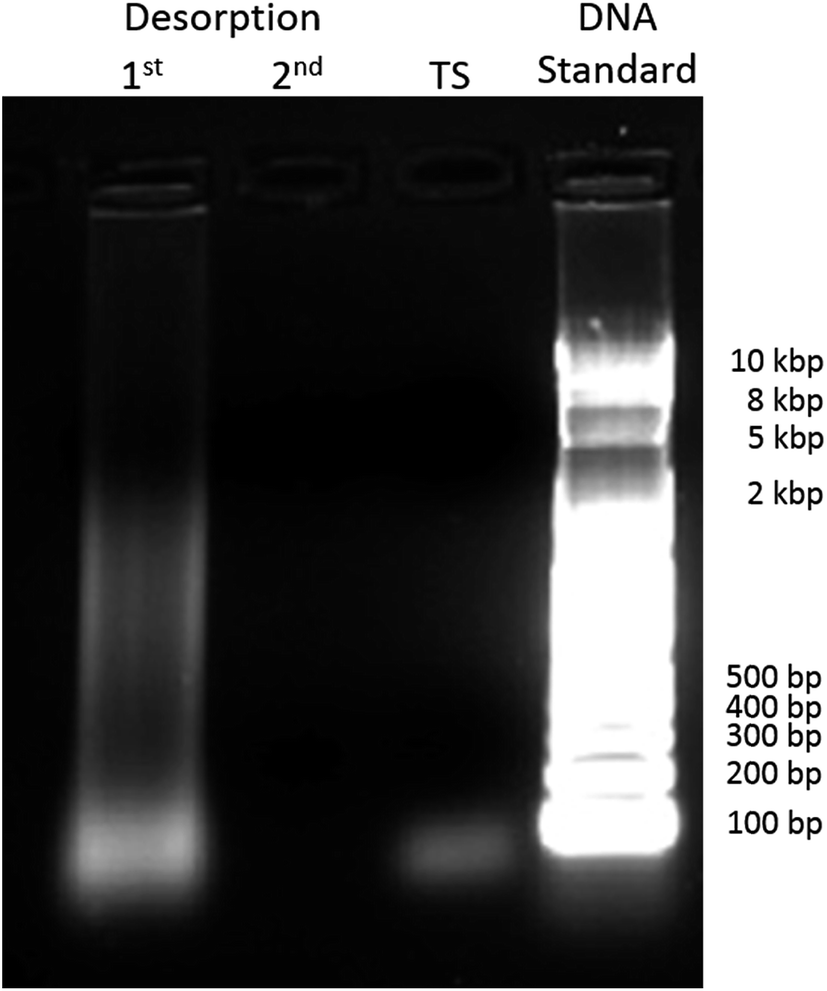 | ||
| Fig. 5 Agarose gel electrophoresis of the non-binding species that were removed after the 1st and 2nd potentiostatic desorption at an applied voltage of 1.3 V between interdigital electrodes for two minutes, together with TS probe control and a 10 kbp DNA standard ladder reference, stained using GelRed. This was done adapting a procedure presented by J. Wang et al.37 | ||
Conclusions
In this work, we designed and built a small and robust biosensor microchip based on two interdigital electrodes and an RTD which were connected through a female USB type-A connector. The RTD was connected to an easy and affordable software and hardware system designed in-house to control the temperature for real-time impedimetric biosensing while incubating the samples. Taking advantage of the telomerase-positive nuclear extract (Jurkat cells) and nuclear lysis buffer, we showed the detection of telomerase activity without the addition of redox, fluorescent labels or other reagents. In under 20 minutes, we obtained up to 14-fold increase in signal when testing a telomerase-positive solution compared to a heat-inactivated telomerase-negative solution, using EIS. We believe that translating this biosensor microchip into a more affordable substrate, with the addition of a simple impedance measurement circuit can lead to an easy and fast cancer detection at the point-of-care.Acknowledgements
This research work was supported in part by NSF-Chemistry Grant no. CHE-1152940 and NASA-URC Grant no. NNX10AQ17A. The authors also want to acknowledge the financial support of the NSF-NSEC Center for Hierarchical Manufacturing Grant no. CHM-CMMI-053117, and the help of John Nicholson at their Conte Nanotechnology Cleanroom Laboratory at the University of Massachusetts, Amherst for the microfabrication of the IDA. We thank the Materials Characterization Center at the University of Puerto Rico, Molecular Sciences Research Building, for the SEM images. L. Cunci acknowledges the Graduate Research Fellowship from NSF-EPSCoR Institute for Functional Nanomaterials (IFN) through Grant no. EPS-1002410. R. Gomez-Moreno and M. Martinez Vargas are supported by NIGMS grant R25GM061838 and A. Baerga-Ortiz is supported by NSF grant CHE-0953254. This work was also supported by grants from the National Institutes of Health to C. I. Gonzalez (GM008102-3052, KO1 HL-04355, U54 CA96297, FIPI, PES-UPR). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.References
- L. Hayflick and P. S. Moorhead, Exp. Cell Res., 1961, 25, 585–621 CrossRef CAS.
- S. B. Cohen, M. E. Graham, G. O. Lovrecz, N. Bache, P. J. Robinson and R. R. Reddel, Science, 2007, 315, 1850–1853 CrossRef CAS PubMed.
- A. J. Cesare and R. R. Reddel, Nat. Rev. Genet., 2010, 11, 319–330 CrossRef CAS PubMed.
- J. Chen, J. Li and Y. Sun, Lab Chip, 2012, 12, 1753–1767 RSC.
- K.-Y. Lien, Y.-H. Chuang, L.-Y. Hung, K.-F. Hsu, W.-W. Lai, C.-L. Ho, C.-Y. Chou and G.-B. Lee, Lab Chip, 2010, 10, 2875–2886 RSC.
- Q. H. Quach, J. Jung, H. Kim and B. H. Chung, Chem. Commun., 2013, 49, 6596–6598 RSC.
- T. Tian, S. Peng, H. Xiao, X. Zhang, S. Guo, S. Wang, X. Zhou, S. Liu and X. Zhou, Chem. Commun., 2013, 49, 2652–2654 RSC.
- H.-b. Wang, S. Wu, X. Chu and R.-Q. Yu, Chem. Commun., 2012, 48, 5916–5918 RSC.
- R. Qian, L. Ding, L. Yan, M. Lin and H. Ju, J. Am. Chem. Soc., 2014, 136, 8205–8208 CrossRef CAS PubMed.
- L. Tian and Y. Weizmann, J. Am. Chem. Soc., 2013, 135, 1661–1664 CrossRef CAS PubMed.
- R. Qian, L. Ding and H. Ju, J. Am. Chem. Soc., 2013, 135, 13282–13285 CrossRef CAS PubMed.
- X. Zhou and D. Xing, Chem. Soc. Rev., 2012, 41, 4643–4656 RSC.
- E. Kulla and E. Katz, Sensors, 2008, 8, 347–369 CrossRef CAS PubMed.
- D. A. Skvortsov, M. E. Zvereva, O. V. Shpanchenko and O. A. Dontsova, Acta Naturae, 2011, 3, 48–68 CAS.
- Y. Xiao, V. Pavlov, T. Niazov, A. Dishon, M. Kotler and I. Willner, J. Am. Chem. Soc., 2004, 126, 7430–7431 CrossRef CAS PubMed.
- U. Eskiocak, D. Ozkan-Ariksoysal, M. Ozsoz and H. A. Oktem, Anal. Chem., 2007, 79, 8807–8811 CrossRef CAS PubMed.
- Z. Shao, Y. Liu, H. Xiao and G. Li, Electrochem. Commun., 2008, 10, 1502–1504 CrossRef CAS PubMed.
- Z. Yi, H.-B. Wang, K. Chen, Q. Gao, H. Tang, R.-Q. Yu and X. Chu, Biosens. Bioelectron., 2014, 53, 310–315 CrossRef CAS PubMed.
- A. Bonanni, C. K. Chua, G. Zhao, Z. Sofer and M. Pumera, ACS Nano, 2012, 6, 8546–8551 CrossRef CAS PubMed.
- L. Wu, J. Wang, L. Feng, J. Ren, W. Wei and X. Qu, Adv. Mater., 2012, 24, 2447–2452 CrossRef CAS PubMed.
- X. Zhou, D. Xing, D. Zhu and L. Jia, Anal. Chem., 2009, 81, 255–261 CrossRef CAS PubMed.
- Y. Li, B. Liu, X. Li and Q. Wei, Biosens. Bioelectron., 2010, 25, 2543–2547 CrossRef CAS PubMed.
- S. Sato and S. Takenaka, Anal. Chem., 2012, 84, 1772–1775 CrossRef CAS PubMed.
- W. Yang, X. Zhu, Q. Liu, Z. Lin, B. Qiu and G. Chen, Chem. Commun., 2011, 47, 3129–3131 RSC.
- Z. Gao, H. Deng, W. Shen and Y. Ren, Anal. Chem., 2013, 85, 1624–1630 CrossRef CAS PubMed.
- A. Bonanni and M. Pumera, ACS Nano, 2011, 5, 2356–2361 CrossRef CAS PubMed.
- S. Guo, D. Wen, Y. Zhai, S. Dong and E. Wang, ACS Nano, 2010, 4, 3959–3968 CrossRef CAS PubMed.
- S. Basuray, S. Senapati, A. Aijian, A. R. Mahon and H. C. Chang, ACS Nano, 2009, 3, 1823–1830 CrossRef CAS PubMed.
- G. A. Koretzky, M. A. Kohmetscher, T. Kadleck and A. Weiss, J. Immunol., 1992, 149, 1138–1142 CAS.
- B. Mari, F. Checler, G. Ponzio, J. F. Peyron, S. Manie, D. Farahifar, B. Rossi and P. Auberger, EMBO J., 1992, 11, 3875–3885 CAS.
- J. X. Pang, X. Y. Chen and S. G. Wu, Diyi Junyi Daxue Xuebao, 2002, 22, 481–485 CAS.
- D. Mukherjee, D. T. Fritz, W. J. Kilpatrick, M. Gao and J. Wilusz, in mRNA Processing and Metabolism, Springer, 2004, pp. 193–211 Search PubMed.
- M. Okada, I. M. Cheeseman, T. Hori, K. Okawa, I. X. McLeod, J. R. Yates, A. Desai and T. Fukagawa, Nat. Cell Biol., 2006, 8, 446–457 CrossRef CAS PubMed.
- N. W. Kim, M. A. Piatyszek, K. R. Prowse, C. B. Harley, M. D. West, P. L. Ho, G. M. Coviello, W. E. Wright, S. L. Weinrich and J. W. Shay, Science, 1994, 266, 2011–2015 CAS.
- W. Zhang, K. Patel, A. Schexnider, S. Banu and A. D. Radadia, ACS Nano, 2014, 8, 1419–1428 CrossRef CAS PubMed.
- S. Borini, R. White, D. Wei, M. Astley, S. Haque, E. Spigone, N. Harris, J. Kivioja and T. Ryhänen, ACS Nano, 2013, 7, 11166–11173 CrossRef CAS PubMed.
- J. Wang, G. Rivas, M. A. Jiang and X. J. Zhang, Langmuir, 1999, 15, 6541–6545 CrossRef CAS.
- L. Sun, Z. Zhang, S. Wang, J. Zhang, H. Li, L. Ren, J. Weng and Q. Zhang, Nanoscale Res. Lett., 2009, 4, 216–220 CrossRef CAS PubMed.
- L. Qingwen, G. Hong, W. Yiming, L. Guoan and M. Jie, Electroanalysis, 2001, 13, 1342–1346 CrossRef.
- I. Feliciano-Ramos, M. Caban-Acevedo, M. Aulice Scibioh and C. R. Cabrera, J. Electroanal. Chem., 2010, 650, 98–104 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09689d |
| This journal is © The Royal Society of Chemistry 2014 |