DOI:
10.1039/C4RA08026B
(Paper)
RSC Adv., 2014,
4, 52349-52356
Highly transparent and flexible silica/cellulose films with a low coefficient of thermal expansion†
Received
2nd August 2014
, Accepted 29th September 2014
First published on 30th September 2014
Abstract
Highly transparent and flexible silica/cellulose films with low thermal expansion coefficients have been prepared by the in situ synthesis of silica in cellulose scaffolds using Na2SiO3 as a precursor. The low content of silica in the composite films had an influence on the tensile strength of the composites. Furthermore, the composite films integrated the merits of cellulose and silica. The transmittance of the composite films in the visible light region were comparable to that of glass, but the films were foldable. Moreover, the composite films had CTEs lower than 15 ppm K−1. The composite films would be the perfect substrates for the future production of electronic devices, such as flexible displays and e-papers, and could replace costly conventional batch processes based on the glass substrates currently used, and eventually be applied to rollable and even foldable devices.
Introduction
Flexible polymer materials with high thermal resistance, transparency, and low thermal expansion properties have attracted significant interest. This kind of material can be used in the emerging field of flexible electronics.1–5 On the other hand, most materials currently used have large coefficients of thermal expansion (CTEs, approx. 50 ppm K−1), and in particular, foldable plastics exhibit extremely large CTEs, in excess of 200 ppm k−1. Moreover, polymers usually have low glass transition temperatures (Tgs) accompanied by abrupt CTE changes, which greatly limit their practical applications in terms of the process temperature. To overcome these limitations, nanocomposites from hybrids of organic polymers and inorganic materials have attracted considerable interest. There have also been many studies on exploring the effects of nanoparticle incorporation with different chemical nature, size and shape on the thermal and optical properties of polymers.6–11
As a new kind of material, biodegradable composites from renewable resources have attracted the attention of researchers in diverse areas. These biohybrids not only display improved structural and functional properties, but also show remarkable biocompatibility, biodegradability and some other novel properties supported by either the biological or inorganic moieties.12–17 Cellulose, which is the most abundant resource on earth, has good mechanical and thermal stability properties at temperatures ranging from room temperature to about 200 °C. Furthermore, it has no Tgs because of the strong inter- and intra-molecular hydrogen bonds. Scaffolds from cellulose nanofibers have been reported to be good matrices for the fabrication of composites with promising thermal stability.18,19 Cellulose nanofibers or nanowhiskers are also good fillers for the preparation of composites with improved thermal properties.20–22 Nevertheless, there are considerable challenges in separating the native cellulose nanofibers from the macroscopic cellulose fibers or developing expensive large-scale fermentation technology for bacterial cellulose. Therefore, the dissolution and regeneration of native cellulose has become an alternative pathway for the preparation of cellulose-based functional materials.
Extensive research has been focused on cellulose dissolution and the construction of functional cellulose materials from the developed solvents. In previous work, aqueous solvents containing alkaline and urea have been developed for cellulose dissolution.23,24 The regenerated cellulose films prepared from LiOH/urea or NaOH/urea aqueous solution had a porous structure, which could be used as scaffolds for the synthesis of inorganic nanoparticles25,26 or curable organic prepolymers27,28 in situ for the construction of functional cellulose materials, and the composites integrated the merits of the cellulose and the incorporated components. Inspired by these interesting characteristics, this study attempted to develop a material with some properties of glass (e.g. high transmittance and low thermal expansion) by the incorporation of silica in a cellulose matrix. The results indicated that the transmittance of the composites was comparable to that of the glass, but they were foldable and had good mechanical properties as well as low CTEs (CTE < 20 ppm k−1). The straightforwardness of the fabrication of the cellulose-based composite films represented not only a scientific advance regarding the identification of compounds of a natural origin and establishing economically efficient routes for the production of biodegradable and/or biocompatible composite materials, but also an emerging area of research aimed at the future design of green bioelectronics.
Experimental section
Chemicals
Native cellulose (Cotton linter, α-cellulose ≥ 95%) was supplied by Hubei Chemical Fiber Co. Ltd. (Xiangfan, China), and its viscosity-average molecular weight (Mη) was about 1.07 × 105, which was determined in cadoxen at 25 °C. The other chemicals were of analytical grade and supplied by the Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). Deionized water was used for the experiments.
Preparation of regenerated cellulose (RC) films and composite films
The freezing–thawing method was used for cellulose dissolution. Briefly, powder-like filter paper (Native cellulose) was dispersed into an aqueous lithium hydroxide/urea solution (4.6 wt%/15.0 wt%), and then placed it in a refrigerator. After it was frozen, the solution was taken out and thawed at room temperature to obtain a transparent cellulose solution (5 wt%). The resultant cellulose solution was subjected to centrifugation at 7500 rpm and 15 °C for 10 min to eliminate bubbles in the viscous solution. The viscous bubble-free solution was cast on a glass plate and the thickness of the solution was controlled to be about 0.9 mm, and immersed it into coagulation bath containing 80 v/v% ethanol to regenerate for 10 min. The regenerated cellulose films (RC) were washed with deionized water and then immersed into a Na2SiO3 solution for 24 h, and then treated with a mixed ethanol and H2SO4 (2 M) solution for 20 min. The composite films were washed with deionized water and dried under ambient conditions. The composite films prepared from the Na2SiO3 solution with a concentration of 2 wt%, 5 wt%, 10 wt%, and 15 wt% was coded as CMS-2, CMS-5, CMS-10, and CMS-15, respectively. The regenerated cellulose film that was not treated with Na2SiO3 solution was coded RC.
Characterization
X-ray diffraction of the regenerated cellulose film and composite films were carried out in reflection mode (Rigaku RINT 2000, Japan) using Ni-filtered CuKa radiation. The Fourier-transform infrared (FT-IR) spectra of the samples were recorded on an FT-IR spectrometer (FT-IR 615, Japan). The samples were ground into powders, mixed with KBr, and pressed to form a sample disk for the FT-IR measurements. For transmission electron microscopy (TEM), the specimens were embedded in a poly (methylmethacrylate–butylmethacrylate) (PMMABMA) resin and then cut with a Leica Ultracut-E using a glass knife. Sections, approximately 100 nm in thickness, were mounted on a grid with a carbon support, and then disembodied by removing the resin with acetone. The section was then examined with a JEOL-1010 apparatus. Scanning electron microscopy (SEM) observations of the surfaces of the RC and composites were carried out using a Hitachi S-4800 microscope. Prior to analysis, the samples were cut into small pieces from the prepared samples and coated with a thin layer of evaporated gold. Thermogravimetric analysis (TGA) was carried out by using an Ulvac TGD 9600. The samples were cut into powders and about 30 mg of the powder was placed into a platinum pan and heated from 20 to 700 °C at a rate of 10 K min−1 in a nitrogen atmosphere. The optical transmittance (Tr) of the cellulose film and composite films were measured with a Shimadzu UVmini-1240 apparatus at wavelengths ranging from 200 to 1000 nm. The mechanical properties of the films were characterized using a tensile tester (CMT 6503, Shenzhen SANS Test machine Co. Ltd, China) according to ASTM/D638-91 at a speed of 5 mm min−1, and five samples were tested for each set of samples. The coefficients of thermal expansion (CTEs) were measured on a thermomechanical analyzer (TMA/SS6000, SII Nanotechnology Inc.). The specimens were 20 mm long and 5 mm wide with a 15 mm span. The measurements were carried out from 30 to 120 °C by elevating the temperature at a rate of 10 °C min−1 in air in tensile mode under a load of 2 g. The linear coefficient of thermal expansion (CTEs) could be expressed as
where ΔL is the change in length of the test specimen due to heating, L0 is the initial length of the test specimen at room temperature, and ΔT is the temperature difference over which the change in the length of the specimen was measured. The CTEs could be obtained from the slope of the curve obtained when the change in film length, ΔL, was represented as a function of the temperature, T. The CTE values were determined in the fourth run.
Results and discussion
The composite films containing silica were prepared by in situ synthesis method, as it was shown in Scheme 1. The RC films displayed a homogeneous macroporous structure, as shown in Fig. 1. This unique structure was due to the phase separation of the cellulose solution during the regeneration process, where solvent-rich regions contributed to pore formation. The N2 adsorption–desorption isotherm of the cellulose hydrogel after being freeze-dried indicated the formation of a continuity of the pore size distribution of macropores, and SBET, which was inferred from the N2 adsorption isotherm by Brunauer–Emmett–Teller analysis of the amount of gas adsorbed at P/P0 between 0.05 and 0.3, was about 270 m2 g−1. When the cellulose hydrogel films were immersed into a Na2SiO3 solution, the solution could be readily impregnated into the cellulose scaffolds through the pores. When the films were treated with a mixed H2SO4 and ethanol aqueous solution, Na2SiO3 transformed immediately into a silica gel, and the transparency of the composite films was improved after the reaction process compared to that of the pristine cellulose films. It must be noted that the composite films were opaque when the cellulose hydrogel films containing Na2SiO3 were treated with ethanol first and then by H2SO4.
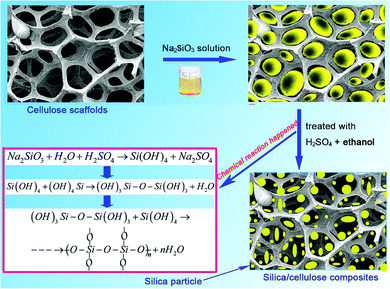 |
| | Scheme 1 Process for the preparation of silica/cellulose composite films. | |
 |
| | Fig. 1 SEM images of the RC film after being freeze-dried, a and b are for the surface and cross-section of the film, respectively. | |
To clarify this interesting phenomenon, the microstructure of the composite films after being freeze-dried were characterized by SEM. Fig. 2 shows the morphologies of the composites after being freeze-dried. The silica components conferred an obvious change in the microstructure of the composites compared to that of the pristine cellulose film. In all samples, obvious macropores had a diameter mainly in the 100 nm range, which was smaller than that of the cellulose film. This suggests that the silica components had an obvious influence on the microstructure of the composites. There was also evidence that all the samples contained an additional mesoporous sub-structure, consisting of fibrils with a diameter of about 30 nm, which was more uniform than that of the cellulose films. This interesting phenomenon was attributed to the contribution of silica components in the cellulose matrix. On the other hand, silica particles were barely detected because the silica sheath was synthesized on the surface of the cellulose nanofibrils. In contrast, in the cellulose hydrogel film containing Na2SiO3 treated with ethanol first and followed by H2SO4, silica particles were formed in the cellulose matrix, see ESI (FS1†). To further clarify the structure of the composite films, TEM was carried out by ultrathin sectioning of resin-embedded composite films, followed by removal of the resin on the TEM grid. Fig. 3 shows the micrographs of the composite films. Silica nanoparticles with a mean particle size of about 20 nm were dispersed uniformly in the cellulose matrix. With increasing Na2SiO3 concentration, the particle size changed slightly, suggesting that the network of the cellulose matrix could act as reaction sites not only for the synthesis of the silica nanoparticles, but also to keep the nanoparticles from growing into large sizes. In the cellulose hydrogel films containing Na2SiO3 treated with ethanol first followed by H2SO4, silica particles with a larger particle size were formed in the cellulose matrix, see ESI (FS1†).
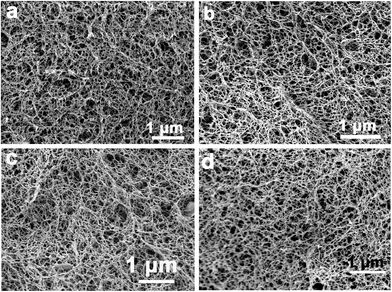 |
| | Fig. 2 SEM images of the silica/cellulose composite films after being freeze-dried, a, b, c, and d represent CMS-2, CMS-5, CMS-10, and CMS-15, respectively. | |
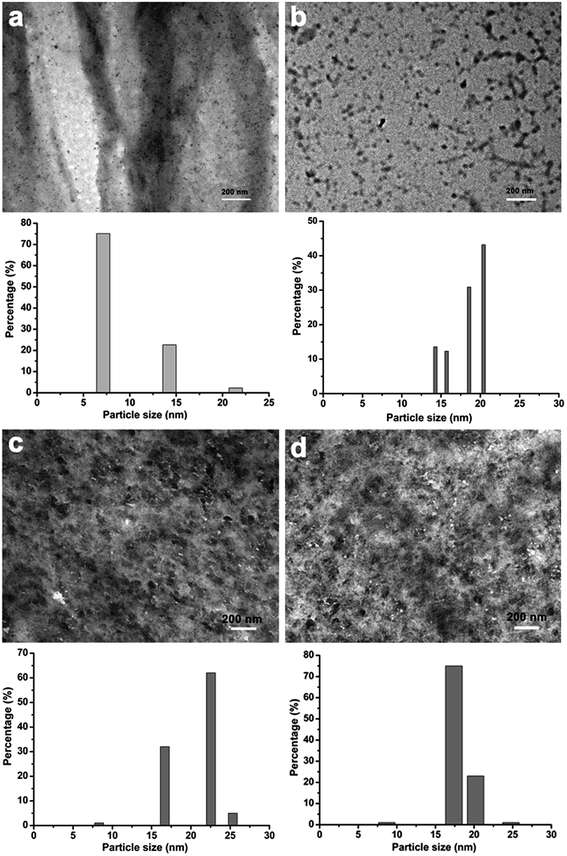 |
| | Fig. 3 Ultrathin-section TEM images of the silica/cellulose composite films and the particle size distribution, the particle size distribution was determined from at least 500 particles, a, b, c, and d represent CMS-2, CMS-5, CMS-10, and CMS-15, respectively. | |
Fig. 4 shows the XRD patterns of the RC and the composite films. The peaks at 12.1, 20.3, and 21.8° 2θ were assigned to the (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0), (110), and (200) crystalline planes of crystalline cellulose II, respectively.29 In addition to the diffraction peaks of cellulose II, there was no diffraction peak for silica observed in the composite films. This suggests that the silica in the composite films was amorphous. The results are consistent with the reported conclusions regarding the preparation of SiO2 from a liquid-phase silica source, such as water glass.30 FT-IR spectroscopy was used to characterize the cellulose and silica/cellulose composite films at the molecular structure level. The spectra of some selected samples are shown in Fig. 5. The strong and broad band centered at ∼3400 cm−1 was assigned to the stretching mode of –OH groups involved in different hydrogen-bonding interactions. They were correlated with –OH of cellulose and molecular water (in the H–O–H deformation region, at ∼1648 cm−1).31 The strongest band with the maximum absorption at ∼1070 cm−1 was assigned to the antisymmetric Si–O–Si stretching mode. The corresponding symmetric mode appeared at ∼800 cm−1. The νas Si–O–Si band was the most informative on the structure of the silica.32
0), (110), and (200) crystalline planes of crystalline cellulose II, respectively.29 In addition to the diffraction peaks of cellulose II, there was no diffraction peak for silica observed in the composite films. This suggests that the silica in the composite films was amorphous. The results are consistent with the reported conclusions regarding the preparation of SiO2 from a liquid-phase silica source, such as water glass.30 FT-IR spectroscopy was used to characterize the cellulose and silica/cellulose composite films at the molecular structure level. The spectra of some selected samples are shown in Fig. 5. The strong and broad band centered at ∼3400 cm−1 was assigned to the stretching mode of –OH groups involved in different hydrogen-bonding interactions. They were correlated with –OH of cellulose and molecular water (in the H–O–H deformation region, at ∼1648 cm−1).31 The strongest band with the maximum absorption at ∼1070 cm−1 was assigned to the antisymmetric Si–O–Si stretching mode. The corresponding symmetric mode appeared at ∼800 cm−1. The νas Si–O–Si band was the most informative on the structure of the silica.32
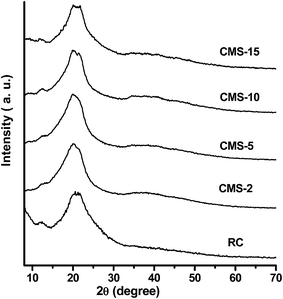 |
| | Fig. 4 XRD patterns of the regenerated cellulose film and composite films. | |
 |
| | Fig. 5 FT-IR spectra of the RC and silica/cellulose composite films. | |
The thermal stability and degradation profile of the RC and composite films was assessed by thermogravimetry, as shown in Fig. 6. A small weight loss of ∼5% around 80 °C was assigned to the release of moisture from the samples. The pure RC films showed two obvious weight loss slopes with elevated temperatures. The first weight loss was found in the temperature range from 300–350 °C, which was attributed to the onset of cellulose decomposition. The second weight loss peak at 420–540 °C was assigned to the decomposition of cellulose. The thermal stability of the composites apparently decreased due to destruction of the microstructure of cellulose. With increasing silica content, the initial decomposition temperature of the composites shifted to lower temperatures. The first weight loss stage for the pure RC film was around 326 °C, which decreased to about 250 °C when silica nanoparticles were loaded, and shifted to lower temperature with increasing level of incorporated silica, which indicated the formation of composites that partially disrupted the interaction in the cellulose matrix. The thermal decomposition temperature of the composite films was higher than those of the synthetic polymers often used in photoelectric devices, such as PMMA (∼170 °C) or PVC (∼200 °C).
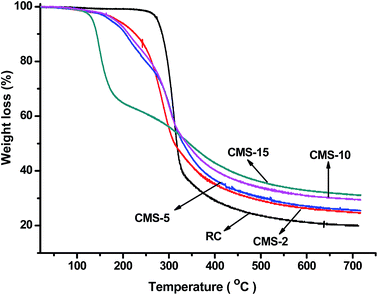 |
| | Fig. 6 Thermogravimetric curves for the RC and silica/cellulose composite films. | |
An extremely important property of the composite films was their improved optical transparency. Fig. 7 shows the optical transmittance spectra in the visible region of the RC and composite films with different silica loadings. The transmittance of the RC films at a 550 nm wavelength was 83%, and the transparency was improved to 92% with a low content of silica nanoparticles. In comparison, the regular transmittance of glass with a thickness of about 1.35 mm is 93% including surface reflection. This suggests that the optical transmittance of the composite films is comparable to that of glass. For a transparent polymer composite with dispersed inorganic fillers, the optical property can be described as
where
χ is the optical path length,
Vp is the particle volume fraction,
r is the particle radius,
λ is the light wavelength, and
np and
nm are the refractive indices of the fillers and matrix, respectively. The difference in the refractive index of the fillers and matrix played an important role in controlling the optical transmittance of the composite films. The refractive index of cellulose and silica are about 1.47 and 1.63, respectively. Therefore, when silica nanoparticles are impregnated into the cellulose matrix, the composite films displays improved transmittance due to the smaller difference in
np and
nm. Nogi
et al. reported that the acetylation of cellulose nanofibers could decrease the refractive index and make it close to the refractive index of resin, and a high transparency composite could be obtained from these two materials. The unique network structure of the RC matrix also contributed to the improvement in the transmittance of the composite films. The porous structure of the cellulose matrix could control the particle size of the silica particles and make their distribution in the cellulose matrix homogeneous, resulting in an increase in the affinity between silica particles and cellulose matrix, which is also related to the high transparency. Because silica particles and cellulose are hydrophilic, the affinity of the silica particles surface and cellulose matrix is increased, allowing the silica particles to form in the cavities of the cellulose matrix tightly. As a result, the scattering of visible light at the interface of the silica particles decreased. The introduction of an excess of silica particles in the cellulose matrix did not result in high transparency of the composite films, but the composite films became brittle.
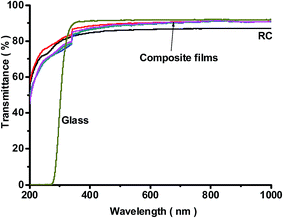 |
| | Fig. 7 Transmittance of the RC and composite films at wavelengths from 200 to 1000 nm. | |
The typical stress–strain curves of the RC and composite films are shown in Fig. 8a. The pure RC film had a tensile strength of about 81 MPa with an elongation at break of about 10.7%, while for the composite films containing a lower content of silica (about 4.5 wt%), the tensile strength of the composite film increased from 81 to 95 MPa, with decreasing elongation at break to about 6%. On the other hand, for the composite films with higher content of silica, the tensile strength decreased from 95 MPa to 86 MPa, but the tensile strength was comparable to that of pure RC. The decrease in the tensile strength of the composite films prepared from different concentrations of silica precursor would be caused by two factors. The first is that the silica content in the composite films increased with increasing concentration of the precursor. A higher silica content would destroy the network structure of the composite films. The second factor could be ascribed the increased particle size of the silica particles. TEM showed that the particles size of silica increased with increasing the precursor concentration. Larger sized silica particles with a higher content could form a stress concentration in the composite. Therefore, the tensile strength decreased with increasing silica filler concentration. The Young's modulus of the composite films increased slightly and then decreased, and there was no obvious difference in the Young's modulus of the composites, as shown in Fig. 8b. Furthermore, the results for composite films demonstrated that unbendable silica materials became ductile by reinforcement with cellulose nanofibrils networks, suggesting that the network structures of cellulose scaffolds suppress crack propagation in the composite films, indicating that the composite films integrated the virtues of silica and cellulose, suppressing break and generating an attractive tensile strength and reliably foldability. Flexibility is an essential characteristic not only for future electronic devices, such as displays and solar cells, but also for materials suitable for roll-to-roll production processes.
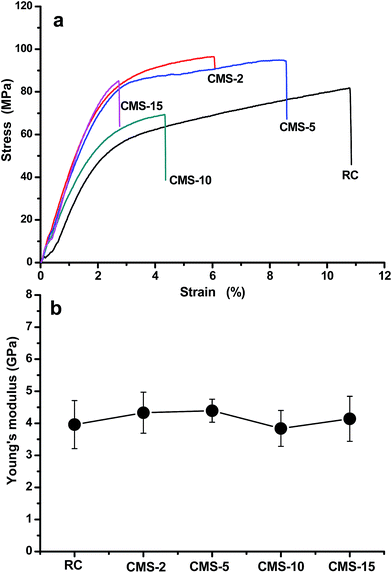 |
| | Fig. 8 Stress–strain curves (a) and Young's modulus of the RC and silica/cellulose composite films prepared from Na2SiO3 solution at different concentrations. | |
The thermal expansion properties of the RC and composite films were investigated using a thermal mechanical analysis chart, the slope of which reflected the coefficient of linear thermal expansion. The obtained length-normalized charts are shown in Fig. 9, and the corresponding CTEs of the samples from the fourth run are shown in Fig. 10. The CTE of the RC films was about 16 ppm K−1 at temperatures ranging from 30 to 100 °C. The CTEs of the composite films was lower than that of the RC film, and it decreased with increasing silica content in the composites. For the CMS-15 composite film with a silica content of about 11.2%, the CTEs were as low as 12 ppm K−1, which was lower than many transparent polymer films current used. Furthermore, the composite film was foldable in this state. The interaction between the polymer matrix and filler surface caused lower CTEs. In our work, the silica nanoparticles were synthesized around the cellulose nanofibrils. When the composites were heated, the silica and cellulose matrix would expand. Silica, however, has lower coefficient of thermal expansion than cellulose. Therefore, the thermal expansion behavior of the cellulose matrix was limited by the silica components. Consequently, the composite films would be difficult to deform by the thermal expansion of the composites. It has been reported that it is important to make a nanofiber network interaction strong enough to restrict the thermal expansion of the matrix. In the current practical application, a transparent film with a low CTE, typically less than 20 ppm K−1, is desirable to match the thermal expansion of the substrates to the deposited OLED layers. This novel property can be explained by the unique network structure of the cellulose film, and the induced thermal stresses that were small enough to be constrained almost completely by the rigid networks of the cellulose matrix. This would be a promising technology for the preparation of advanced cellulose composite films for functional applications.
 |
| | Fig. 9 Thermal expansion curves of the RC and silica/cellulose composite films at temperatures ranging from 30 to 100 °C, the data used for the figure was obtained from the fourth run. | |
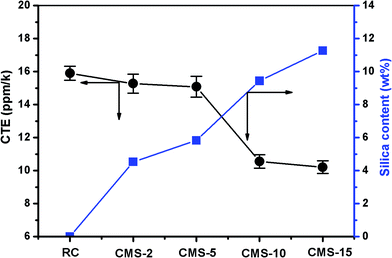 |
| | Fig. 10 Relationship between the silica content and the coefficient of thermal expansion of the RC and composite films. The CTE values were determined in the fourth run. | |
Conclusions
In summary, flexible, transparent composite films with structural robustness were prepared successfully using a facile process. The composite films integrated the merits of the cellulose and silica, and they were mechanically strong, flexible and with obvious low CTEs. The incorporated silica had little influence on the thermal properties and an obvious influence on the mechanical properties of the composite films, and the composite films had high optical transmittance; the transmittance at 550 nm was higher than 80%. These results suggest that the prepared composite films potentially have a wide range of practical applications owing to their desirable properties, including mechanical flexibility, optical transparency and mechanical robustness, as well as low CTEs properties.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 51273085 and 51003043), and the project by the Fundamental Research Funds for the Central Universities (2014PY024).
References
- M. C. Choi, Y. Kim and C. S. Ha, Prog. Polym. Sci., 2008, 33, 581–630 CrossRef CAS PubMed.
- D. Ghosh, P. Ghosh, M. Tanemura, A. Haysahi, Y. Hayashi, K. Shinji, N. Miura, M. Z. Yusop and T. Asaka, Chem. Commun., 2011, 47, 4980–4982 RSC.
- B. H. Lee, S. H. Park, H. Back and K. Lee, Adv. Funct. Mater., 2011, 21, 487–493 CrossRef CAS.
- W. A. MacDonald, J. Mater. Chem., 2004, 14, 4–10 RSC.
- L. C. Tomé, R. J. Pinto, E. Trovatti, C. S. Freire, A. J. Silvestre, C. P. Neto and A. Gandini, Green Chem., 2011, 13, 419–427 RSC.
- H. Althues, J. Henle and S. Kaskel, Chem. Soc. Rev., 2007, 36, 1454–1465 RSC.
- J. Opatkiewicz, M. C. LeMieux and Z. Bao, ACS Nano, 2010, 4, 2975–2978 CrossRef CAS PubMed.
- I. Perez-Baena, I. Loinaz, D. Padro, I. García, H. J. Grande and I. Odriozola, J. Mater. Chem., 2010, 20, 6916–6922 RSC.
- Y. Rao and T. N. Blanton, Macromolecules, 2008, 41, 935–941 CrossRef CAS.
- G. Wu, H. Xu and T. Zhou, Polymer, 2010, 51, 3560–3567 CrossRef CAS PubMed.
- Y. Yin and C. Wang, J. Sol-Gel Sci. Technol., 2011, 59, 36–42 CrossRef CAS.
- H. Fukuzumi, T. Saito, T. Iwata, Y. Kumamoto and A. Isogai, Biomacromolecules, 2008, 10, 162–165 CrossRef PubMed.
- S. Ifuku, S. Morooka, A. N. Nakagaito, M. Morimoto and H. Saimoto, Green Chem., 2011, 13, 1708–1711 RSC.
- M. Nogi and H. Yano, Adv. Mater., 2008, 20, 1849–1852 CrossRef CAS.
- M. Nogi and H. Yano, Appl. Phys. Lett., 2009, 94, 233 CrossRef PubMed.
- M. Nogi, S. Iwamoto, A. N. Nakagaito and H. Yano, Adv. Mater., 2009, 21, 1595–1598 CrossRef CAS.
- L. Carro, E. Hablot and T. Coradin, J. Sol-Gel Sci. Technol., 2014, 70, 263–271 CrossRef CAS.
- C. Y. Yan, J. Wang, W. Kang, M. Cui, X. Wang, C. Y. Foo, K. J. Chee and P. S. Lee, Adv. Mater., 2014, 26, 2022–2027 CrossRef CAS PubMed.
- Z. Fang, H. Zhu, Y. Yuan, D. Ha, S. Zhu, C. Preston, Q. Chen, Y. Li, X. Han, S. Lee, G. Chen, T. Li, J. Munday, J. Huang and L. Hu, Nano Lett., 2014, 14, 765–773 CrossRef CAS PubMed.
- N. Ucar, O. Ayaz, E. Bahar, Y. Wang, M. Oksuz, A. Onen, M. Ucar and A. Demir, Text. Res. J., 2013, 83, 1335–1344 CrossRef PubMed.
- M. F. Rosa, E. S. Medeiros, J. A. Malmonge, K. S. Gregorski, D. F. Wood, L. H. C. Mattoso, G. Glenn, W. J. Orts and S. H. Imam, Carbohydr. Polym., 2010, 81, 83–92 CrossRef CAS PubMed.
- A. J. Svagan, M. A. S. Azizi Samir and L. A. Berglund, Biomacromolecules, 2007, 8, 2556–2563 CrossRef CAS PubMed.
- S. Liu and L. Zhang, Cellulose, 2009, 16, 189–198 CrossRef CAS.
- J. Zhou, S. Liu, J. Qi and L. Zhang, J. Appl. Polym. Sci., 2006, 101, 3600–3608 CrossRef CAS.
- S. Liu, J. Zhou and L. Zhang, J. Phys. Chem. C, 2011, 115, 3602–3611 CAS.
- S. Liu, Q. Yan, D. Tao, T. Yu and X. Liu, Carbohydr. Polym., 2012, 89, 551–557 CrossRef CAS PubMed.
- S. Liu, D. Tao, T. Yu, H. Shu, R. Liu and X. Liu, Cellulose, 2013, 20, 907–918 CrossRef CAS.
- W. Li, Y. Wu, W. Liang, B. Li and S. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 5726–5734 CAS.
- L. Zhang, D. Ruan and J. Zhou, Ind. Eng. Chem. Res., 2001, 40, 5923–5928 CrossRef CAS.
- S. L. Pan, J. J. Zhang and G. Z. Song, Mater. Sci. Technol., 2009, 25, 1437–1441 CrossRef CAS PubMed.
- C. J. Lee, G. S. Kim and S. H. J. Hyun, Mater. Sci., 2002, 37, 2237–2241 CrossRef CAS.
- A. Fidalgo and L. M. J. Ilharco, J. Non-Cryst. Solids, 2011, 283, 144–154 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra08026b |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 












