
Actinide elements and germanium: a first-principles density functional theory investigation of the electronic and magnetic properties of ApGe (Ap = Ac–Lr) diatoms
Abstract
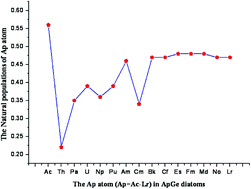
The geometry and electronic and magnetic properties of ApGe (Ap = Ac–Lr) diatoms have been studied using first-principles density functional theory, with relativistic effects being taken into account. The calculated natural populations of ApGe diatoms show that the electronic charge is transferred mainly from Ap to Ge, most of the Ap 5f subshell in ApGe being inert and not involved in chemical bonding. The calculated highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) gaps of the ApGe diatoms exhibit an oscillating behavior from AcGe to BkGe, and a slight increase from CfGe to NoGe. The calculated magnetic moments of ApGe show that the total magnetic moment depends mainly on the 5f electrons of Ap in the ApGe diatoms, which generate the magnetic properties. Our calculated results are in good agreement with the published theoretical and experimental data.
 Please wait while we load your content...
Please wait while we load your content...

