DOI:
10.1039/C4RA09579K
(Paper)
RSC Adv., 2014,
4, 55716-55722
2,4-Dinitrobenzenesulfonic acid in an efficient Brønsted acid-catalyzed controlled/living ring-opening polymerization of ε-caprolactone†
Received
1st September 2014
, Accepted 14th October 2014
First published on 14th October 2014
Abstract
The ring-opening polymerization of ε-caprolactone (ε-CL) using benzyl alcohol (BnOH) as initiator and 2,4-dinitrobenzenesulfonic acid (DNBA) as catalyst in acetonitrile at room temperature with a [ε-CL]0/[BnOH]0/[DNBA]0 ratio of 40/1/1 has been investigated. The polymerization proceeded to obtain poly(ε-caprolactone) (PCL) with controlled molecular weights. In addition, 1H NMR, SEC, and MALDI-ToF MS measurements demonstrated the initiator residue at the polymer chain end. Furthermore, propargyl alcohol, 5-hexen-1-ol, 2-hydroxyethyl methacrylate, 1,3-propanediol, and pentaerythritol were used as functional initiators to successfully obtain end-functionalized and α,ω-dihydroxy telechelic polyesters. The block copolymerization of PCL and PVL or PTMC provided conditions to afford well-defined poly(ε-caprolactone)-block-poly(δ-valerolactone) (PCL-b-PVL) and poly(ε-caprolactone)-block-poly(trimethylene carbonate) (PCL-b-PTMC).
Introduction
Aliphatic polyesters are attractive biodegradable and biocompatible polymers used in pharmaceuticals and medicine.1–4 In the past, polyesters were mainly prepared by ring-opening polymerization (ROP) using metal-based catalysts;5–11 however, residual metals limited their wider applications. Consequently, much effort was focused on metal-free catalysis. Metal-free catalysts, called organocatalysts, are developing rapidly.1,12 In particular, using organocatalysts to synthesize well-controlled macromolecular architectures has been studied extensively. Since the first report in 2001, Hedrick and Waymouth have used 4-dimethylaminopyridine13 for living ROP of lactide. Later, various organic base catalysts were developed, such as N-heterocyclic carbene,14–17 thiourea/amine,18,19 guanidine,20 phosphazene,21,22 1,5,7-triazabicyclo [4.4.0] dec-5-ene,23,24 and 1,8-diazabicyclo[5.4.0]-undec-7-ene.25,26 the ROP of cyclic esters.
Comparatively, Brønsted acid catalysts have not been widely reported. Pioneered in 2000 by Endo and co-workers using HCl·Et2O-catalyzed in ROP of lactones,27,28 acid catalysts such as methanesulfonic acid,29,30 trifluoromethane sulfonic acid,29,31 diphenyl phosphate,32–34 and triflimide,35,36 catalyze the ROP of cyclic esters and have attracted more and more attention. To further extend the scope of the acid catalysis system, our group reported several efficient acid catalysts for the controlled/living ROP of cyclic esters.37–40
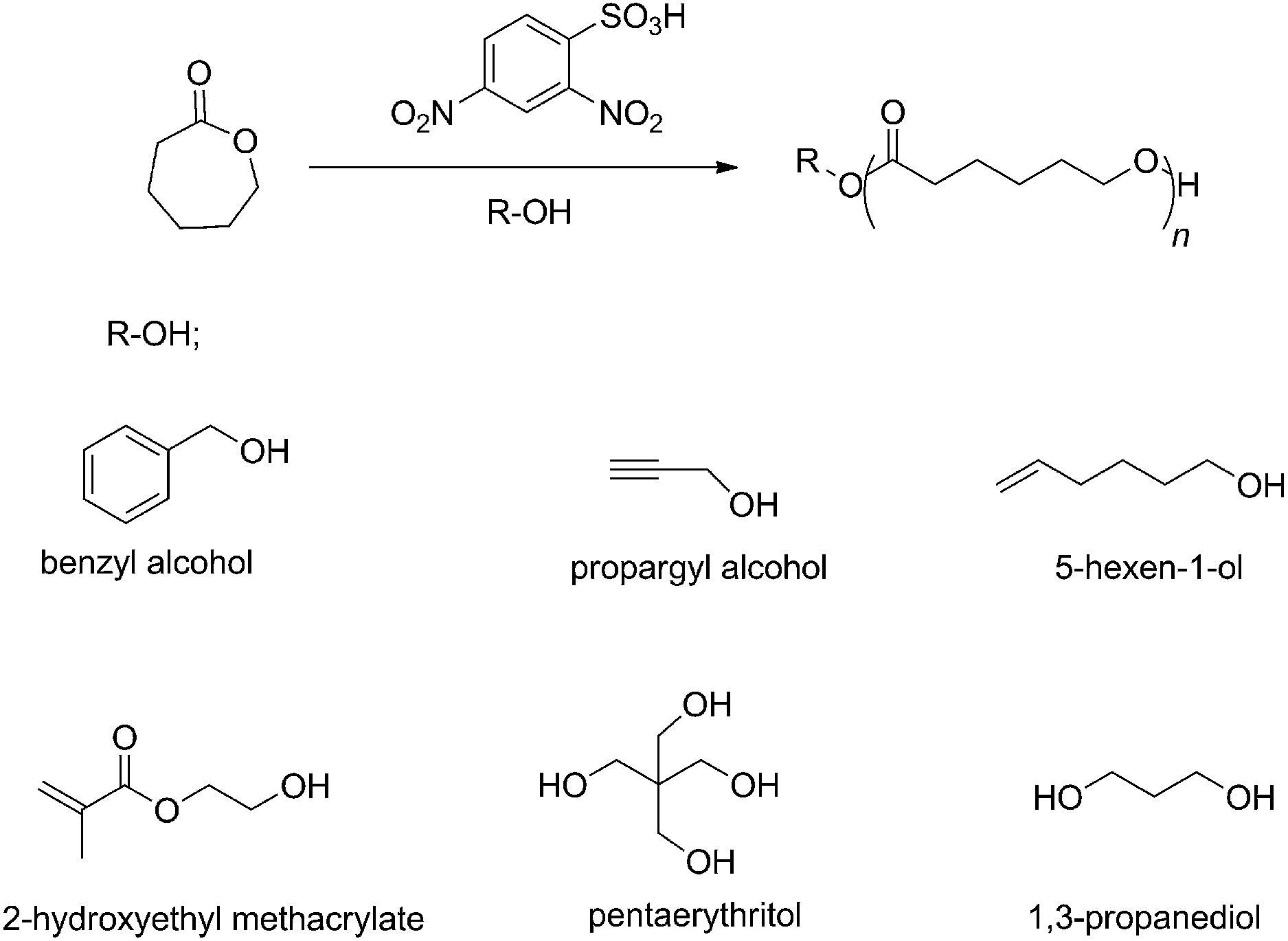
Previously, efficient catalytic performance in Hosomi–Sakurai,41,42 Ritter,43 Friedel–Crafts,44 and enantioselective protonation45 reactions was shown for 2,4-dinitrobenzenesulfonic acid (DNBA) as Brønsted acid. Additionally, DNBA is commercially available, inexpensive, low in toxicity, and easy to store and handle. As reported by Córdova in 2004,46 the advantages of organic catalysts are low cost and low toxicity, but the ROP of CL required high temperature (120 °C). In contrast, the ROP in the current study was conducted at room temperature. In accordance with the excellent properties of DBNA, we decided to evaluate it as catalyst in the ROP of ε-CL. DNBA is a powerful organocatalyst for controlled/living ROP of ε-CL, which promoted rapid polymerization. A strong electron-withdrawing group strengthens the acidity of Brønsted acids;47 the structural components of DNBA comprise a benzene ring connected with two nitro-groups and a sulfonic group. It is likely the strong electron-withdrawing group (NO2) that provides the organic acid catalyst with high reactivity. To date, as far as we know, DNBA has not yet been applied in ROP. In this article, we describe (1) the characterization and optimization of DNBA for controlled ROP of ε-caprolactone, (2) the controlled/living nature of the DNBA-catalyzed ROP of ε-CL by 1H NMR, SEC, and MALDI-ToF MS analyses, (3) various initiators used to initiate the polymerizations and produce end-functionalized and α,ω-dihydroxy telechelic polymers (as shown in Scheme 1), and (4) the synthesis of diblock copolymers consisting of PCL with PVL or PTMC.
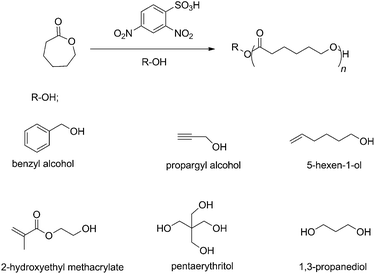 |
| | Scheme 1 Ring-opening polymerization (ROP) of ε-caprolactone (ε-CL) using 2,4-dinitrobenzenesulfonic acid (DNBA) as the organocatalyst and various alcohols (R–OH) as initiators. | |
Experimental
Materials
Both ε-caprolactone (ε-CL; 99%, Sinopharm Chemical Reagent) and δ-valerolactone (δ-VL; 99%, Sinopharm Chemical Reagent) were distilled over CaH2 in an inert environment. Trimethylene carbonate (TMC) was synthesized via a conventional method.48 Acetonitrile (CH3CN; >99% water content, <0.001%) was dried over 3 Å molecular sieve pellets for 48 h before use. Benzyl alcohol (BnOH; 99%, Acros) was refluxed over CaH2 prior to distillation. DNBA (Tokyo Kasei Kogyo Co., Ltd. (TCI)) was dried prior to use. Both 5-hexen-1-ol (98%, Energy Chemical) and 2-hydroxyethyl methacrylate (99.5%, J&K Scientific Co.) were distilled under reduced pressure before use. Next, 1,3-propanediol (99%, Arocs) was azeotropically distilled with toluene. Pentaerythritol (99%, Arocs) was dried over P2O5 under high vacuum. Propargyl alcohol (99%, Alfa Aesar) was distilled over CaH2. Triethylamine (98%, Sinopharm Chemical Reagent Co.) was used as received.
Characterizations
The number-average molecular weight (Mn,NMR) and monomer conversion were determined from the 1H NMR spectra in CDCl3 on a Bruker ARX-250 spectrometer at 300 MHz at ambient temperature. Size exclusion chromatography (SEC) was performed in tetrohydrofuran (THF) using an SSI 1500 pump equipped with Waters column (5 μm, 300 × 7.8 mm) at a flow rate of 0.7 mL min−1 at 25 °C, and a Wyatt Optilab rEX differential refractive index (DRI) detector with a 658 nm light source. Polystyrene was used as the standard to determine the number-average molecular weight (Mn) and molecular weight distributions (Mw/Mn) of the polymers. All SEC data were processed using Wyatt Astra V 6.1.1 software. Matrix-assisted, laser desorption/ionization time-of-flight mass spectra (MALDI-ToF MS) was performed using a mass spectrometer (ultraflextreme; Bruker Co.) with Smartbeam/Smartbeam II modified Nd:YAG laser. The MALDI-ToF mass spectra represent averages over 500 laser shots at a 25 kV acceleration voltage. The polymer sample was dissolved in CHCl3 at a concentration of 5 mg mL−1; the matrix 2,5-DHB (2,5-dihydroxybenzoic acid) was dissolved in aqueous (1%, 10 μL) solution of trifluoroacetic acid and acetonitrile (volume ratio = 70/30).
General procedure for polymerization of ε-caprolactone
All reactions were conducted in a glove box at room temperature. The ε-caprolactone (ε-CL) (0.213 mL, 2 mmol, 40 equiv.) was mixed into acetonitrile ([ε-CL]0 = 1.0 mol L−1) using benzyl alcohol (5.2 μl, 0.05 mmol, 1 equiv.) as the initiator. DNBA (0.0124 g, 0.05 mmol, 1 equiv.) was then dissolved in the reaction solutions. After 4 h, the monomer conversion reached 95.2%, which was determined by 1H NMR. At the end of the polymerization, an excess of triethylamine was added to stop the reaction. The polymer was dissolved in the minimum amount of dichloromethane and separated out from cold methanol, and then dried in a vacuum drying oven. Conversion follows: 95.2%, yield, 61.0%; 1H NMR (CDCl3): δ (ppm), 1.39 (m, 2H × n, (–CH2CH2CH2CH2CH2–)n), 1.63 (m, 2H × n, (–CH2CH2CH2O–)n), 1.68 (m, 2H × n, (–COCH2CH2CH2–)n), 2.31 (t, 2H × n, J = 7.3 Hz, (–OCOCH2CH2–)n), 3.65 (t, 2H, J = 6.6 Hz, CH2CH2OH), 4.06 (t, 2H × n, J = 6.6 Hz, (–CH2CH2O–)n), 5.12 (s, 2H, ArCH2O), 7.23–7.39 (m, 5H, aromatic); Mn,NMR ≈ 4580 g mol−1, Mw/Mn = 1.16.
Block copolymerization of ε-caprolactone and δ-valerolactone or trimethylene carbonate
All reactions were conducted in a glove box and at room temperature. The ε-caprolactone (ε-CL) (0.213 mL, 2 mmol, 40 equiv.) was mixed into acetonitrile ([ε-CL]0 = 1.0 mol L−1). Benzyl alcohol (5.2 μl, 0.05 mmol, 1 equiv.) was used as the initiator. The DNBA (0.0124 g, 0.05 mmol 1 equiv.) was then dissolved in the reaction solutions. After 6 h (we extended the reaction time for full conversion of monomers), 40 equiv. of δ-VL (0.181 mL, 2 mmol) was then added to the reaction chamber, and start the block copolymerization to obtain PCL-b-PVL under the same conditions. An excess of triethylamine was used to quench the reaction. The polymer was dissolved in a small quantity of CH2Cl2 and isolated by cold methanol. Yield, 53%. 1H NMR (CDCl3): δ (ppm), 1.38 (m, 2H × n, (–CH2CH2CH2CH2CH2–)n), 1.63 (m, 2H × (n + m), (–CH2CH2CH2O–)n), 1.68 (m, 2H × n, (–COCH2CH2CH2–)n), 2.31 (t, 2H × (n + m), J = 7.3 Hz, (–COCH2CH2–)n), 3.65 (t, 2H, J = 6.5Hz, –CH2CH2OH), 4.07 (t, 2H × (n + m), J = 6.6 Hz, (–CH2CH2O–)n), 5.12 (s, 2H, ArCH2O), 7.23–7.39 (m, 5H, aromatic). Mn,NMR ≈ 8140 g mol−1, Mw/Mn = 1.16.
Block copolymerization of ε-caprolactone and trimethylene carbonate (PCL-b-PTMC) was carried out with the similar reaction conditions. Yield, 57.0%. 1H NMR (CDCl3): δ (ppm), 1.39 (m, 2H × n, (–CH2CH2CH2CH2CH2–)n), 1.66 (m, 4H × n, (–COCH2CH2CH2–)n), 1.93 (n, 2H, (–OCH2CH2OH)), 2.01 (m, 2H × m, (–OCH2CH2–)m), 2.31 (t, 2H × n, J = 6.8 Hz, (–COCH2–)n), 3.75 (m, 2H, –CH2OH), 4.07 (m, 2H × n, (–OCH2CH2CH2O–)n), 4.25 (t, 4H × m, J = 6.4 Hz, (–OCH2CH2CH2O–)m), 5.12 (s, 2H, ArCH2O), 7.24–7.38 (m, 5H, aromatic). SEC (THF): Mn,NMR ≈ 8480 g mol−1, Mw/Mn = 1.15.
Results and discussion
Ring-opening polymerization of ε-caprolactone using benzyl alcohol and 2,4-dinitrobenzenesulfonic acid
In order to evaluate the activity of 2,4-dinitrobenzenesulfonic acid, we used DNBA as the catalyst and benzyl alcohol as the initiator in the ROP of ε-caprolactone in CH3CN (1.0 mol L−1) at room temperature with [ε-CL]0/[BnOH]0/[DNBA]0 = 40/1/1 (Table 1, Run 4). The monomer conversion reached 95.2% after 4 h, which was determined from the 1H NMR spectrum. To find the optimization of DNBA-catalyzed ROP of ε-CL reaction conditions, different solvents were carried out in DNBA-catalyzed ROP of ε-CL. (Table 1, Run 1–4). Toluene and THF were used as the solvent, while no polymer was separated out; thus, they acted as poor solvents for DNBA-catalyzed ROP of ε-CL. DNBA could be partly soluble in CH2Cl2 (completely dissolved at the end of polymerization); thus moderate monomer conversion (45.7% after 12 h) took place. The results demonstrated that CH3CN was a suitable solvent for DNBA-catalyzed ROP of ε-CL. In this paper, we report experiments for ROP of CL using other organocatalysts (diphenyl phosphate (DPP), triflimide (HNTf2) and trifluoromethanesulfonic acid (HOTf)) from other catalytic systems to compare catalytic performance, at room temperature with the [M]0/[I]0/[C]0 ratio of 40/1/1 to afford PCL, as shown in Table 1 (Runs 4–6). All results demonstrated that 2,4-dinitrobenzenesulfonic acid (DNBA) was superior for the ROP of ε-CL.
Table 1 ROP of ε-caprolactone (ε-CL) catalyzed by 2,4-dinitrobenzenesulfonic acid (DNBA) with benzyl alcohol (BnOH) as the initiatora
| Run |
[M]/[I] |
Solvent |
Time (h) |
Conv.b (%) |
Mn,calcdc (g mol−1) |
Mn,NMRb (g mol−1) |
Mw/Mnd |
Mwe (g mol−1) |
| [M]0 = 1.0 mol L−1; room temperature. Determined by 1H NMR in CDCl3. Calculated from ([M]0/[BnOH]0) × conv. × (Mw of ε-CL) + (Mw of BnOH). Determined by SEC in THF using polystyrene standards. Determined by SEC in THF using polystyrene standards and correction factors.30,49,50 Catalyzed by diphenyl phosphate (DPP); [M]0 = 1.0 mol L−1; room temperature; in toluene; [M]0/[I]0/[C]0 = 40/1/1. Catalyzed by triflimide (HNTf2); [M]0 = 1.0 mol L−1; in CH2Cl2; [M]0/[I]0/[C]0 = 40/1/1. Catalyzed by trifluoromethanesulfonic acid (HOTf); [M]0 = 1.0 mol L−1; in CH2Cl2; [M]0/[I]0/[C]0 = 40/1/1. |
| 1 |
40 |
CH2Cl2 |
12 |
45.7 |
2190 |
— |
— |
|
| 2 |
40 |
Toluene |
12 |
0 |
— |
— |
— |
|
| 3 |
40 |
THF |
12 |
0 |
— |
— |
— |
|
| 4f |
40 |
Toluene |
8 |
96.3 |
4500 |
4460 |
1.10 |
2509 |
| 5g |
40 |
CH2Cl2 |
12 |
87.3 |
4100 |
4330 |
1.16 |
3472 |
| 6h |
40 |
CH2Cl2 |
7 |
96.7 |
4520 |
4260 |
1.21 |
3978 |
| 7 |
40 |
CH3CN |
4 |
95.2 |
4450 |
4580 |
1.16 |
4231 |
| 8 |
80 |
CH3CN |
8 |
97.9 |
9050 |
9190 |
1.12 |
6673 |
| 9 |
100 |
CH3CN |
14 |
92.1 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 620 620 |
11180 |
1.14 |
9979 |
| 10 |
160 |
CH3CN |
20 |
89.0 |
16360 |
16580 |
1.11 |
3978 |
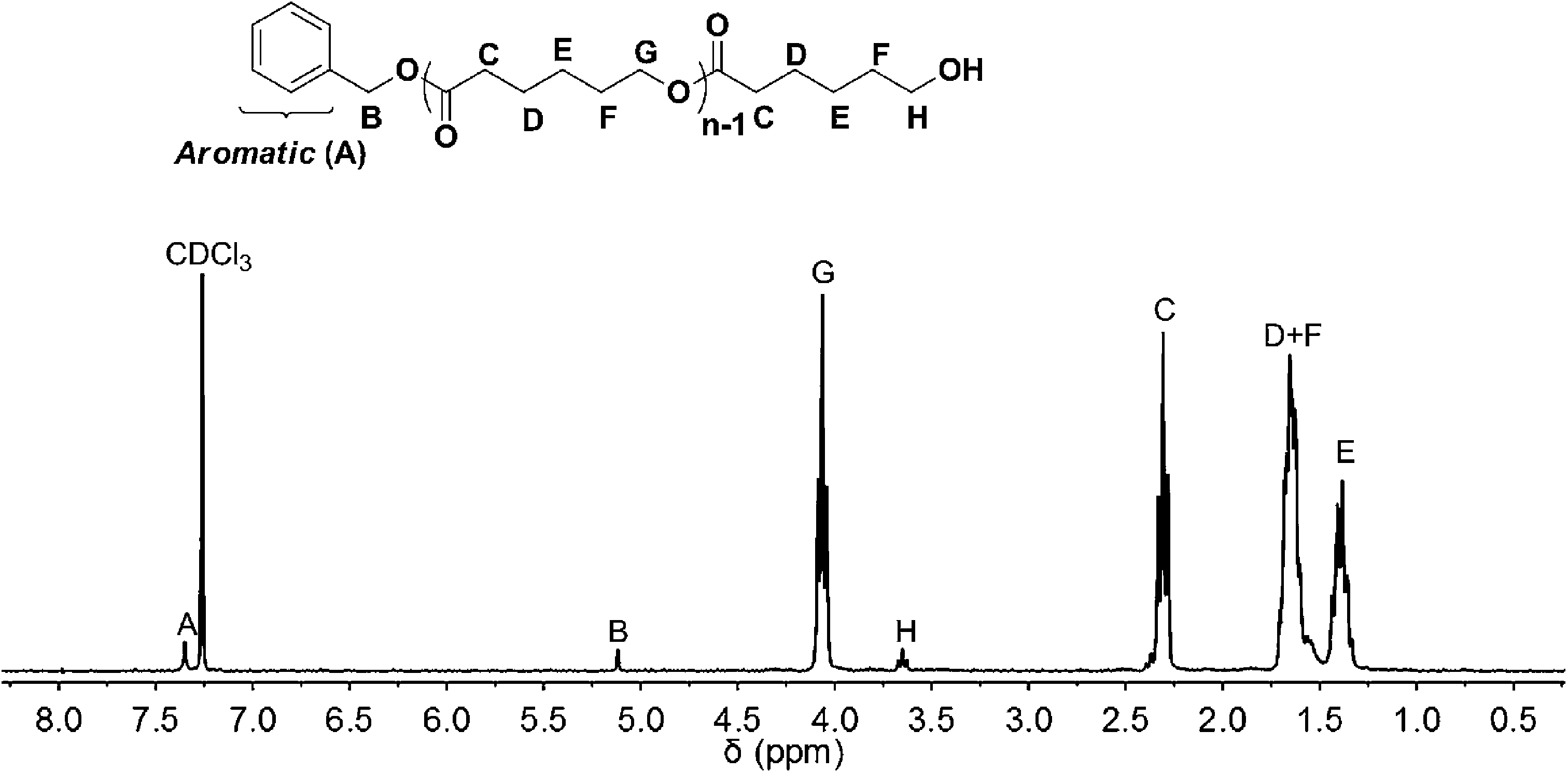
According to 1H NMR spectra of the obtained PCL (Fig. 1), the peaks of initiator (BnOH) were observed; they appeared in the range of 7.23–7.39 ppm (A) and 5.12 ppm (B). The peaks for the polymer chain (PCL) were observed in 2.31 (C), 1.62–1.70 (D + F), 1.39 (E), 4.05 (G), and 3.65 (H) ppm, respectively. These results demonstrated that the obtained PCL was initiated from BnOH.
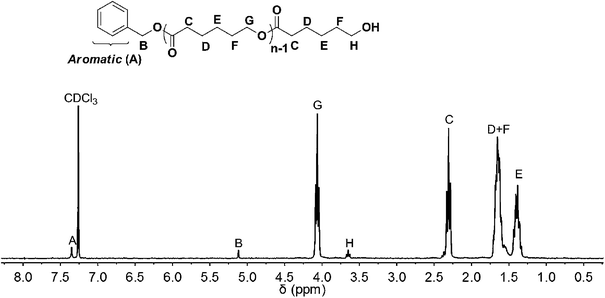 |
| | Fig. 1 1H NMR spectrum (CDCl3, 300 MHz) of poly(ε-caprolactone) ([ε-CL]0/[BnOH]0/[DNBA]0 = 40/1/1) CH3CN, rt, [M]0 = 1.0 mol L−1, 4 h. | |
Moreover, in order to confirm the controlled/living nature of the polymerizations, we carried out ROPs of ε-CL by varying the [ε-CL]0/[BnOH]0 ratio from 40 to 160 (Table 1, Runs 4–7). The results showed that the obtained PCLs of predicted Mn (NMR) agreed with calculated ones by the initial ratio of [ε-CL]0/[BnOH]0 and the monomer conversions. For example, in the 40-mer (Table 1, Run 4), the values of 4580 g mol−1 detected by 1H NMR were a near match to calculated values of 4450 g mol−1. In addition, molecular weights of polymers were higher than 16000 g mol−1, with simultaneous polydispersity (Mw/Mn) values of PCLs ranging from 1.19 to 1.12 via SEC analysis.
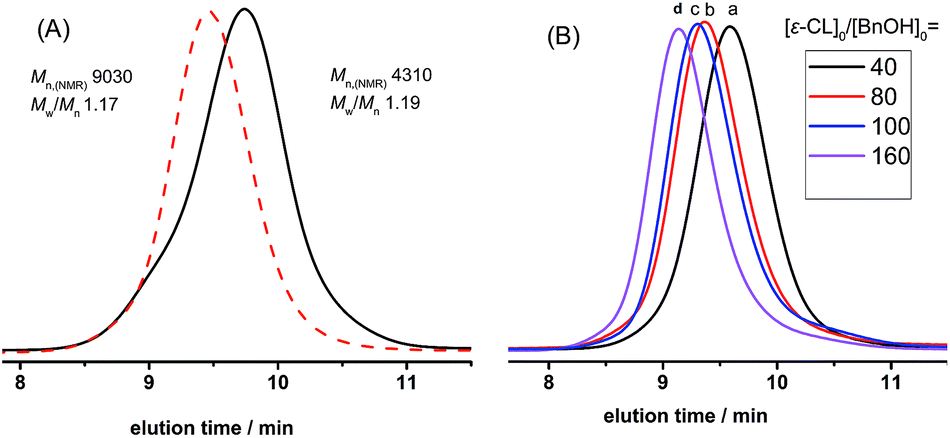
Furthermore, the kinetics and postpolymerization experiments provided convincing evidence of DNBA-catalyzed ROP of ε-CL in controlled/living nature. As shown in Fig. 2, we explored [ε-CL]0 = 1.0 mol L−1 and [ε-CL]0/[BnOH]0/[DNBA]0 = 40/1/1; a linear relationship between monomer conversion and molecule mass values of PCLs was observed (Fig. 2(A)). The plots of ln([M]0/[M]) versus reaction time also fit a strict linear relationship (Fig. 2(B)). Fig. 3(A) shows the SEC traces for chain extension experiments; the first polymerization proceeded with [ε-CL]0/[BnOH]0/[DNBA]0 = 40/1/1 in CH3CN at room temperature without quenching, the monomer conversion reached 97% after 6 h, and PCL with Mn,NMR = 4310 g mol−1, Mw/Mn = 1.19. Additional ε-CL (40 equiv.) was added to the second polymerization, obtaining PCL with Mn,NMR = 9030 g mol−1, Mw/Mn = 1.17, indicating the chain end group of PCL with living nature. Next, Fig. 3(B) shows the SEC traces of varying [ε-CL]0/[BnOH]0 from 40 to 160, and PCLs with low polydispersities. All results indicated characteristics of controlled/living nature.
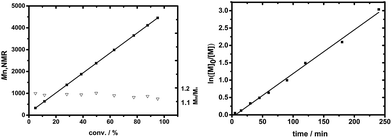 |
| | Fig. 2 (A) Molecular weight (Mn) and polydispersity (Mw/Mn) versus the monomer conversion of ε-CL (theoretical Mn (solid line)) calculated from 108.13 (Mw of BnOH) + conv. × ([M]/[I]) × 114.07 (Mw of ε-CL). (B) Kinetic plots for the polymerization of ε-CL ([ε-CL]0/[BnOH]0/[DNBA]0 = 40/1/1). | |
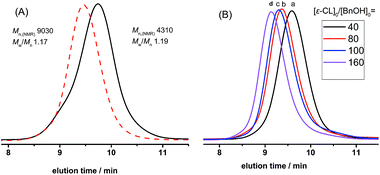 |
| | Fig. 3 (A) SEC traces of the first PCL sequence (solid line) and postpolymerization (dashed line). (B) SEC traces of PCLs with various [M]0/[I]0 ratios of 40 (a), 80 (b), 100 (c), 160 (d). Eluant, THF; flow rate, 0.7 mL min−1. | |
In order to provide further evidence, MALDI-ToF MS measurements were undertaken to prove that the DNBA-catalyzed ROP of ε-CL was initiated by BnOH. As shown in Fig. 4, the MALDI-ToF MS analyses of PCL with monomers to initiator ratios of 40, in which PCL had a molecular formula of molar mass M = 108.13 (Mw of BnOH) + n × 114.07 (Mw of ε-CL) + 23 (Na+) or M = 108.13 (Mw of BnOH) + n × 114.07 (Mw of ε-CL) + 39 (K+), indicating the BnOH residue at the chain end. In addition, the mass differences between two adjacent peaks comprised a ε-CL unit. Those results strongly implied that BnOH initiated the polymerization, and emerged in a living manner without backbiting, transesterification or other undesirable side reactions. In addition, although the monomer (ε-CL), the catalyst (2,4-dinitrobenzenesulfonic acid), the initiator (BnOH) and the solvent (acetonitrile) were subjected to a rigorous drying process before use, concomitant loss of H2O also occurred. From the MALDI-ToF MS spectrum, another two series of undesired peaks were initiated by H2O. Obviously, measured values of water-initiated peaks were equal to theoretical values (as shown in Fig. 4). According to a report by Penczek and co-workers, the Brønsted acid-catalyzed ROP of cyclic esters is an activated monomer mechanism (AM) leading to well-defined polyesters;51 we have proposed a similar AM mechanism as shown in Scheme 2.
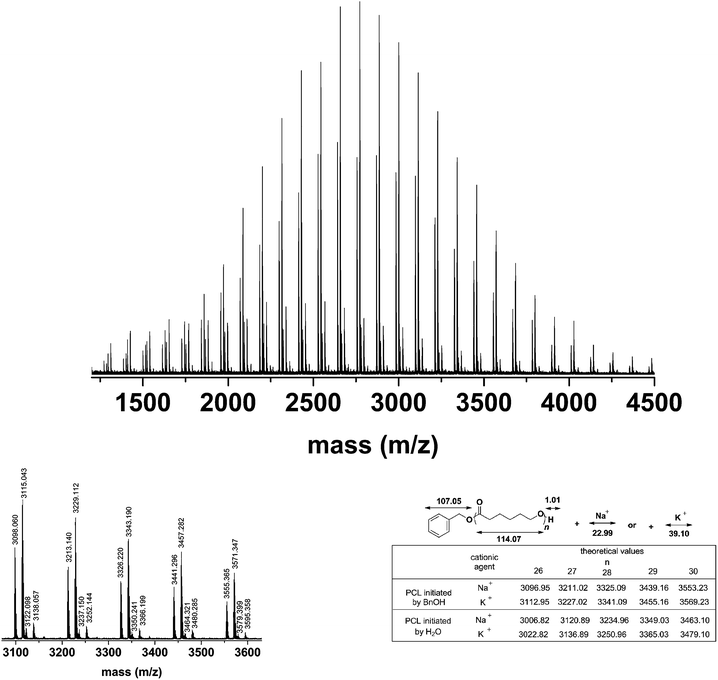 |
| | Fig. 4 MALDI-ToF MS spectra of the obtained PCL ([ε-CL]0/[BnOH]0/[DNBA]0 = 40/1/1, conversion = 95.2%, Mn,NMR = 4580 g mol−1, Mw/Mn = 1.16). | |
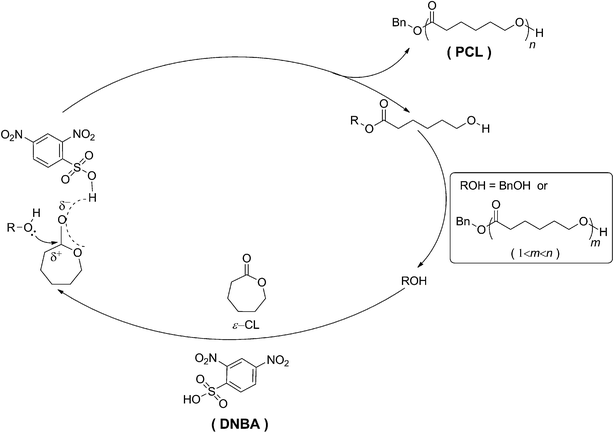 |
| | Scheme 2 Activated monomer mechanism for DNBA-catalyzed ROP of ε-CL using BnOH as the initiator. | |
Syntheses of end-functionalized and α,ω-dihydroxy telechelic poly(ε-caprolactone)
For the purposes of evaluating DNBA-catalyzed polymerizations with the controlled/living nature characteristic, we used DNBA-catalyzed ROP of ε-CL with propargyl alcohol, 5-hexen-1-ol, 2-hydroxyethyl methacrylate, 1,3-propanediol, and pentaerythritol as initiators for providing end-functionalized and α,ω-dihydroxy telechelic polymers, which were confirmed by 1H NMR spectra (Fig. S1–S5†). The obtained polymers with the alkyne group could serve for the click reaction,52,53 and polymers with the methacrylate group as macromonomers, and polymers with alkene group could be used for further modifications.52,54 Table 2 lists the results for obtained poly(ε-caprolactone), and the PCLs initiated by propargyl alcohol, 5-hexen-1-ol, 2-hydroxyethyl methacrylate, pentaerythritol, and 1,3-propanediol; the values of Mn,NMR of 4390, 4250, 4410, 4490 and 4460 g mol−1 correspond to the Mn,calcd of 4510, 4540, 4610, 4190 and 4620 g mol−1, respectively. Meanwhile, the Mw/Mn was at the range of 1.10–1.17 (Table 2), and showed narrow distributions. Thus, we demonstrated that DNBA was an efficient organocatalyst to synthesize well-defined macromolecular architectures.
Table 2 Syntheses of end-functionalized and α,ω-dihydroxy telechelic poly(ε-caprolactone) by the DNBA-catalyzed ROP of ε-CL using various initiatorsa
| Run |
Initiator |
Conv.b (%) |
Mn,calcdc (g mol−1) |
Mn,NMRb (g mol−1) |
Mw/Mnd |
Mwe |
| At room temperature; solvent, CH3CN; time, 10 h; [M]0/[I]0/[DNBA]0 = 40/1/1; [M]0 = 1.0 mol L−1. Determined by 1H NMR in CDCl3. Calculated from ([M]0/[I]0) × conv. × (Mw of ε-CL) + (Mw of initiator). Determined by SEC in THF using polystyrene standards. Determined by SEC in THF using polystyrene standards and correction factors.30,49,50 HEMA = 2-hydroxyethyl methacrylate. |
| 1 |
Propargyl alcohol |
96.7 |
4510 |
4390 |
1.11 |
2497 |
| 2 |
5-Hexen-1-Ol |
97.2 |
4540 |
4250 |
1.13 |
2531 |
| 3 |
HEMAf |
98.1 |
4610 |
4410 |
1.15 |
4119 |
| 4 |
Pentaerythritol |
98.3 |
4190 |
4490 |
1.17 |
3763 |
| 5 |
1,3-Propanediol |
90.1 |
4620 |
4460 |
1.10 |
2508 |
Synthesis diblock copolymers of ε-caprolactone and δ-valerolactone or trimethylene carbonate
According to the chain extension experiments, we concluded that the chain end of obtained PCL had a living nature. Indeed, we also explored the copolymerization of ε-CL with two different monomers (δ-VL and TMC) producing PCL-b-PVL and PCL-b-PTMC, which were versatile as biodegradable materials, and it also illustrated that the hydroxyl group at the chain end possessed further polymerization ability. We carried out the first polymerization with [ε-CL]0/[BnOH]0/[DNBA]0 = 40/1/1 after the monomer was consumed completely without quenching, and then added another monomer (δ-VL, 40 equiv.) for the second polymerization. In addition, the reaction with TMC (40 equiv.), the second monomer to synthesize diblock copolymers (PCL-b-PTMC), was carried out with the same method. The obtained copolymers were determined by 1H NMR spectra (ESI, Fig. S6 and S7†). The SEC traces then illustrated that the molar mass of the obtained PCL-b-PVL shifted from 4270 to 8140 g mol−1, and the polydispersity was slightly narrowed from 1.18 to 1.16; and the molar mass of PCL-b-PTMC shifted from 4560 to 8480 g mol−1, and the polydispersity varied from 1.16 to 1.15, as shown in Fig. 5. All results indicated that DNBA was an efficient catalyst in synthesizing well-defined diblock copolymers of ε-CL with δ-VL and TMC.
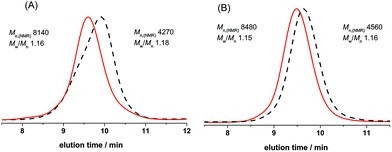 |
| | Fig. 5 (A) SEC traces of first sequence of poly(ε-caprolactone) (PCL; dashed line) and poly(ε-caprolactone)-block-poly(δ-valerolactone) (PCL-b-PVL; solid line). (B) SEC traces of first sequence of poly(ε-caprolactone) (PCL; dashed line) and poly(ε-caprolactone)-block-poly(trimethylene carbonate) (PCL-b-PTMC; solid line). | |
Conclusions
In this work, we used DNBA as an efficient Brønsted acid organocatalyst for ROP of ε-caprolactone (ε-CL) and BnOH as the initiator to produce well-defined poly(ε-caprolactone) (PCL). The controlled/living nature of the polymerization was confirmed, and the obtained polymers demonstrated narrow polydispersity even at high molecular weights. Moreover, we used various initiators (propargyl alcohol, 5-hexen-1-ol, 2-hydroxyethyl methacrylate, 1,3-propanediol, pentaerythritol) to produce end-functionalized and α,ω-dihydroxy telechelic PCL. We also synthesized well-defined block copolymers of PCL-b-PVL and PCL-b-PTMC. DNBA as a commercially available, inexpensive and shelf-stable acidic catalyst may be applicable in broad range of ROPs; we will extend DNBA catalysis in diverse cyclic esters.
Acknowledgements
This work was supported by the National High Technology Research and Development Program of China (2011AA02A202); Doctoral Program of Higher Education of China (20123221110009); and Priority Academic Program Development, Jiangsu Higher Education Institutions. We would like to thank Dr Hailong Liu of Nanjing Normal University for conducting the MALDI-TOF MS experiments and Dr Zhe Song of China Pharmaceutical University for NMR analyses.
Notes and references
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- D. G. Barrett and M. N. Yousaf, Molecules, 2009, 14, 4022–4050 CrossRef CAS PubMed.
- P. Lecomte and C. Jerome, in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Kunkel, G. W. Coates, R. Reichardt, E. Dinjus andT. A. Zevaco, 2012, vol. 245, pp. 173–217 Search PubMed.
- H. R. Kricheldorf, Chem. Rev., 2009, 109, 5579–5594 CrossRef CAS PubMed.
- A. P. Dove, Chem. Commun., 2008, 6446–6470 RSC.
- H. R. Kricheldorf, I. Kreiser-Saunders and A. Stricker, Macromolecules, 2000, 33, 702–709 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed.
- J. C. Wu, T. L. Yu, C. T. Chen and C. C. Lin, Coord. Chem. Rev., 2006, 250, 602–626 CrossRef CAS PubMed.
- C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832–4846 RSC.
- A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC.
- D. Bourissou, S. Moebs-Sanchez and B. Martin-Vaca, C. R. Chim., 2007, 10, 775–794 CrossRef CAS PubMed.
- F. Nederberg, E. F. Connor, M. Moller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712–2715 CrossRef CAS.
- E. F. Connor, G. W. Nyce, M. Myers, A. Mock and J. L. Hedrick, J. Am. Chem. Soc., 2002, 124, 914–915 CrossRef CAS PubMed.
- G. W. Nyce, T. Glauser, E. F. Connor, A. Mock, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2003, 125, 3046–3056 CrossRef CAS PubMed.
- A. P. Dove, H. Li, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2006, 2881–2883 RSC.
- N. E. Kamber, W. Jeong, S. Gonzalez, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2009, 42, 1634–1639 CrossRef CAS.
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 13798–13799 CrossRef CAS PubMed.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863–7871 CrossRef CAS.
- L. Zhang, R. C. Pratt, F. Nederberg, H. W. Horn, J. E. Rice, R. M. Waymouth, C. G. Wade and J. L. Hedrick, Macromolecules, 2010, 43, 1660–1664 CrossRef CAS.
- L. Zhang, F. Nederberg, J. M. Messman, R. C. Pratt, J. L. Hedrick and C. G. Wade, J. Am. Chem. Soc., 2007, 129, 12610–12611 CrossRef CAS PubMed.
- L. Zhang, F. Nederberg, R. C. Pratt, R. M. Waymouth, J. L. Hedrick and C. G. Wade, Macromolecules, 2007, 40, 4154–4158 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- A. Chuma, H. W. Horn, W. C. Swope, R. C. Pratt, L. Zhang, B. G. G. Lohmeijer, C. G. Wade, R. M. Waymouth, J. L. Hedrick and J. E. Rice, J. Am. Chem. Soc., 2008, 130, 6749–6754 CrossRef CAS PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- H. A. Brown, A. G. De Crisci, J. L. Hedrick and R. M. Waymouth, ACS Macro Lett., 2012, 1, 1113–1115 CrossRef CAS.
- Y. Shibasaki, H. Sanada, M. Yokoi, F. Sanda and T. Endo, Macromolecules, 2000, 33, 4316–4320 CrossRef CAS.
- F. Sanda, H. Sanada, Y. Shibasaki and T. Endo, Macromolecules, 2002, 35, 680–683 CrossRef CAS.
- S. Gazeau-Bureau, D. Delcroix, B. Martin-Vaca, D. Bonrissou, C. Navarro and S. Magnet, Macromolecules, 2008, 41, 3782–3784 CrossRef CAS.
- D. Delcroix, B. Martin-Vaca, D. Bourissou and C. Navarro, Macromolecules, 2010, 43, 8828–8835 CrossRef CAS.
- D. Bourissou, B. Martin-Vaca, A. Dumitrescu, M. Graullier and F. Lacombe, Macromolecules, 2005, 38, 9993–9998 CrossRef CAS.
- K. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999–2005 CrossRef CAS.
- K. Makiguchi, Y. Ogasawara, S. Kikuchi, T. Satoh and T. Kakuchi, Macromolecules, 2013, 46, 1772–1782 CrossRef CAS.
- D. Delcroix, A. Couffin, N. Susperregui, C. Navarro, L. Maron, B. Martin-Vaca and D. Bourissou, Polym. Chem., 2011, 2, 2249–2256 RSC.
- R. Kakuchi, Y. Tsuji, K. Chiba, K. Fuchise, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2010, 43, 7090–7094 CrossRef CAS.
- K. Makiguchi, S. Kikuchi, T. Satoh and T. Kakuchi, J. Polym. Sci. Part A: Polym. Chem., 2013, 51, 2455–2463 CrossRef CAS.
- S. Kan, Y. Jin, X. He, J. Chen, H. Wu, P. Ouyang, K. Guo and Z. Li, Polym. Chem., 2013, 4, 5432 RSC.
- J. Chen, S. Kan, H. Xia, F. Zhou, X. Chen, X. Jiang, K. Guo and Z. Li, Polymer, 2013, 54, 4177–4182 CrossRef CAS PubMed.
- X. He, Y. Ji, Y. Jin, S. Kan, H. Xia, J. Chen, B. Liang, H. Wu, K. Guo and Z. Li, J. Polym. Sci. Part A: Polym. Chem., 2014, 52, 1009–1019 CrossRef CAS.
- Y. Jin, Y. Ji, X. He, S. Kan, H. Xia, B. Liang, J. Chen, H. Wu, K. Guo and Z. Li, Polym. Chem., 2014, 5, 3098–3106 RSC.
- D. Kampen, A. Ladepeche, G. Classen and B. List, Adv. Synth. Catal., 2008, 350, 962–966 CrossRef CAS.
- D. Kampen and B. List, Synlett, 2006, 2589–2592 CAS.
- R. Sanz, A. Martinez, V. Guilarte, J. M. Alvarez-Gutierrez and F. Rodriguez, Eur. J. Org. Chem., 2007, 4642–4645 CrossRef CAS.
- W. Zhou, L.-W. Xu, L. Li, L. Yang and C.-G. Xia, Eur. J. Org. Chem., 2006, 5225–5227 CrossRef CAS.
- E. M. Beck, A. M. Hyde and E. N. Jacobsen, Org. Lett., 2011, 13, 4260–4263 CrossRef CAS PubMed.
- J. Casas, P. V. Persson, T. Iversen and A. Cordova, Adv. Synth. Catal., 2004, 346, 1087–1089 CrossRef CAS.
- C. H. Cheon and H. Yamamoto, Chem. Commun., 2011, 47, 3043–3056 RSC.
- J. Matsuo, K. Aoki, F. Sanda and T. Endo, Macromolecules, 1998, 31, 4432–4438 CrossRef CAS.
- M. Save, M. Schappacher and A. Soum, Macromol. Chem. Phys., 2002, 203, 889–899 CrossRef CAS.
- I. Palard, M. Schappacher, B. Belloncle, A. Soum and S. M. Guillaume, Chem. - Eur. J., 2007, 13, 1511–1521 CrossRef CAS PubMed.
- S. Penczek, J. Polym. Sci. Part A: Polym. Chem., 2000, 38, 1919–1933 CrossRef CAS.
- H. Misaka, R. Kakuchi, C. Zhang, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2009, 42, 5091–5096 CrossRef CAS.
- J. N. Hoskins and S. M. Grayson, Macromolecules, 2009, 42, 6406–6413 CrossRef CAS.
- M. Xie, J. Shi, L. Ding, J. Li, H. Han and Y. Zhang, J. Polym. Sci. Part A: Polym. Chem., 2009, 47, 3022–3033 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR for end-functionalized and α,ω-dihydroxy telechelic poly(ε-caprolactone) and block copolymerization; and calculation details of ε-CL conversion. See DOI: 10.1039/c4ra09579k |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Xu Zhi,
Cheng Chen,
Chengxu Zhao,
Xiaopei Li,
Qiguo Zhang and
Kai Guo*
,
Xu Zhi,
Cheng Chen,
Chengxu Zhao,
Xiaopei Li,
Qiguo Zhang and
Kai Guo*