
Transient nature of graphene quantum dot formation via a hydrothermal reaction†
Takashi Ogi*a,
Hideharu Iwasakiab,
Kana Aishimaa,
Ferry Iskandarc,
Wei-Ning Wangd,
Kazuo Takimiyae and
Kikuo Okuyamaa
aDepartment of Chemical Engineering, Graduate School of Engineering, Hiroshima University, 1-4-1 Kagamiyama, Higashi-Hiroshima 739-8527, Japan. E-mail: ogit@hiroshima-u.ac.jp
bBattery Materials Research Laboratory, Kurashiki Research Center, Kuraray, Co., Ltd., 2045-1 Sakazu, Kurashiki, Okayama 710-0801, Japan
cDepartment of Physics, Institute of Technology Bandung, Ganesha 10, Bandung 40132, West Java, Indonesia
dDepartment of Mechanical and Nuclear Engineering, Virginia Commonwealth University, 401 West Main Street, Richmond, Virginia 23284, USA
eEmergent Molecular Function Research Group, RIKEN Center for Emergent Matter Science (CEMS), Hirosawa 2-1 Wako, Saitama 351-0198, Japan
First published on 21st October 2014
Abstract
A facile, economic and environmentally friendly one-step approach for the preparation of highly luminescent graphene quantum dots (GQDs) was developed using a hydrothermal reaction between citric acid and urea. Unlike previous reports, we focused on the effect of the transient nature of GQD formation on the photoluminescence (PL) properties and molecular structure changes of the products. We found that the GQDs have an optimum reaction time and require an effective precursor to achieve excellent luminescent properties. The PL, ultraviolet-visible (UV-vis) absorption, zeta potential, and nuclear magnetic resonance (NMR) analyses of the GQDs prepared at various reaction times revealed that the molecular structures responsible for the luminescence of the GQDs are aggregates or condensation products of citric acid amides. We found that urea addition to the precursor drastically enhances the PL intensity of the GQDs, and it is 40 times higher than those prepared using the pure citric acid precursor. Additionally, a GQDs–polyvinyl alcohol composite achieved an excellent quantum yield (QY) of 43.6%.
Introduction
Carbon-related luminescent materials including carbon nanotubes (CNTs),1 nanodiamonds,2 carbon dots (C-dots),3 boron carbon oxynitride (BCNO),4,5 and aluminium boron carbon oxynitride (AlBCNO)6 have attracted growing interest in recent years because of their unique properties such as low toxicity, high luminescence, chemical inertness and cost-effective preparation. These luminescent carbon materials show great promise in a wide range of applications such as bioimaging,7,8 photocatalysis,9 electrocatalysis,10 sensing,11 light emitting diodes (LEDs),12 and energy conversion or energy storage devices.13Emerging luminescent carbon nanomaterial, graphene quantum dots (GQDs),14,15 has recently attracted an increasing amount of attention due to its numerous novel chemical/physical properties.14,16 GQDs can be prepared by several physical or chemical techniques such as acidic oxidation,17 hydrothermal synthesis,14 microwave method,18 ultrasonic chemistry synthesis,19 electrochemical method,20 oxygen plasma etching,21 stepwise solution chemistry,13,22 catalysed cage-opening,23 pyrolysis and exfoliation.24 Most of these methods require surface passivation reagents and the reported quantum yields (QY) for GQDs without surface passivation are relatively low.15 Additionally, these methods usually involve tedious processes, expensive starting materials or harsh synthetic conditions, which severely limit the availability of large quantities of luminescent carbon nanomaterials for practical applications. Therefore, the development of simple, convenient, cost-effective and environmentally friendly approaches for the large scale synthesis of luminescent GQDs with a high QY is still a challenge.
Recently, hydrothermal reactions have attracted attention as a simple, low-cost, one-step synthesis route for GQDs. Tang et al. firstly reported a simple method for the preparation of GQDs by microwave-assisted hydrothermal method using glucose.25 The prepared GQDs show excellent luminescence properties and the QY of the GQDs was about 7–11% without surface passivation agents or inorganic additives. Wu et al. synthesized GQDs via a one-pot hydrothermal method using citric acid and dicyandiamide as starting materials at 180 °C and at a constant reaction time of 180 min.26 The as-prepared GQDs have excellent photoluminescence (PL) properties with a maximum QY of 36.5%, and they also have high photo-stability in complex systems.
While, Dong et al. reported a simple bottom-up method to selectively prepare GQDs and graphene oxide (GO) by tuning the degree of carbonization of citric acid.27 In this method, the GQDs and GO were prepared by directly pyrolyzing citric acid at 200 °C. When we focus on the reaction process of GQDs formation, the color of the liquid sample changed from colorless to pale yellow, and then to orange in 30 min, and finally a black solid was obtained after about 120 min. From their detailed analyses, they concluded that GQDs formed in the orange solution and that GO formed in a black solid state. The QYs of the GQDs and the GO at 365 nm were about 9.0% and 2.2%, respectively. Therefore, this research suggests that the focusing of transient nature in GQDs formation in each reaction stage is a crucial for the synthesis of highly luminescent GQDs. However, to the best of our knowledge the evaluation of transient nature of GQDs formation on the luminescence properties and chemical structures changes of the product materials has never been reported.
Herein, we thus investigated for the first time the transient nature of GQDs formation by examining the time-dependence of PL, PL excitation (PLE), ultraviolet-visible (UV-vis) absorption, and chemical structure formation. In this work, citric acid and urea were used as precursors for the synthesis of GQDs by a hydrothermal method. Our method does not require expensive reagents, strong acids, bases, organic solvents and further surface passivation. Moreover, less reaction time is required compared with previously reported methods. Ninety minutes is sufficient for the synthesis of highly luminescent GQDs. The prepared GQDs exhibit excellent water-solubility and a high QY. Additionally, a possible formation mechanism of highly luminescent GQDs is also proposed based on nuclear magnetic resonance (NMR) and Fourier transform infrared spectroscopy (FTIR) analyses of the samples prepared at various reaction times. Furthermore, the GQDs easily mixed with a water-soluble polymer (polyvinyl alcohol: PVA) and this GQDs–PVA composite material shows homogeneous emission and a highly luminescent QY as well, indicating their potential application in the polymer composite material field.
Experimental
Material and synthesis
To examine the time-dependence of sample characterization, we developed the experimental apparatus of hydrothermal synthesis as shown in ESI (Fig. S1†). The experimental apparatus is designed in such a way that sampling tube mounted on reactor withdraws the liquid samples by the internal pressure expeditiously. In this work, citric acid and urea were used as precursors for the synthesis of GQDs. The synthetic procedure are shown in ESI (Fig. S2(a)†). The precursor solutions were prepared by dissolving citric acid (CA) (C6O8H7, SIGMA-ALDRICH Co., Ltd., St. Louis, MO, USA) and urea ((NH2)2CO, SIGMA-ALDRICH Co., Ltd., St. Louis, MO, USA) in pure water at 25 °C for 10 min. The concentration of citric acid and urea in the precursor solutions were fixed at 0.0042 mol L−1 and 0.25 mol L−1, respectively.The pH of the starting solution was 3.0. The precursor solution was then transferred into a 50 mL stirred type stainless steel autoclave and heated at 180 °C and at 0.8–1.0 MPa for 600 min. To obtain the time course of GQDs formation, an aliquot of the solution sample was periodically withdrawn and analysed for its optical and physical properties without further treatment like surface passivation. Fig. S2(b)† shows the relationship between reaction time and the corresponding temperature of the solution samples in the reactor vessel. The temperature of the solution increased as the reaction time increased and a maximum temperature of 180 °C was reached.
To produce the GQDs–PVA composite materials, 10 mg polyvinyl alcohol 500 ((C2H4O)n, KANTO CHEMICAL Co., Ltd., Tokyo, Japan) was dissolved in a 100 mL mixed solution of isopropyl alcohol (C3H5O, Kishida Chemicals Co., Ltd., Osaka, Japan) and water (isopropyl alcohol/water = 3 [g g−1]) at 130 °C for 60 min, and then 1 g aqueous GQDs was mixed with the PVA aqueous solution. The obtained solution was kept at 130 °C for 60 min and cooled to room temperature.
Materials characterization
PL and PLE spectra were measured using a spectrofluorophotometer (RF 5300PC, Shimadzu Corp., Kyoto, Japan) equipped with a xenon lamp. The internal QY was measured using an absolute PL quantum yield measurement system (C9920 02, Hamamatsu Photonics, Shizuoka, Japan) with a BaSO4 coated integrating sphere and a xenon lamp as the light source. All PL analyses were performed at room temperature under excitation at 365 nm, unless otherwise stated. UV-vis absorption spectra of the samples were obtained using an absorption spectrophotometer (UV-2450, Shimadzu Corp.). Elemental and functional groups analysis was performed on an X-ray photoelectron spectrometer (XPS) (Quantera II, PHI Corp.) and a Fourier transform infrared spectrometer (FTIR) (IRAffinity-1S, Shimadzu Corp.), respectively. The effect of reaction time on the zeta potential of the GQDs was measured using a Zetasizer (ZETASIZER NANO, Malvern Instruments Ltd., Worcestershire, UK). All NMR spectra were acquired on a Bruker Avance 600 spectrometer equipped with a triple-resonance CryoProbe at 25 °C. The chemical shifts were observed using a series of two-dimensional (2D) 1H–13C heteronuclear single-quantum coherence (HSQC) spectra. The morphology of the produced samples was observed by transmission electron microscopy (TEM) (JEM-3000F, JEOL, Tokyo, Japan) at an accelerating voltage of 300 kV.Results and discussion
The time course of the reaction for the precursor solution was monitored by digital photography, PL, PLE, UV-vis absorption, and zeta potential measurements. Fig. 1(a) shows the colors of the liquid samples produced by the hydrothermal reaction. The initial liquid sample was transparent (colorless) and it changed to pale orange over 90 min, and it became deep orange as time progressed. Finally, the liquid sample turned black upon 600 min of reaction time. From the digital photographs of the samples exposed to 365 nm excitation, blue emission could be observed by the naked eye for the samples collected over 72 min. The corresponding PL and PLE spectra are shown in Fig. 1(b) and (c). For the samples obtained before 90 min, emission bands centred around 460 nm were observed under excitation at 365 nm. The emission intensity increased as the reaction time increased, and it reached a maximum PL intensity at 90 min, after which the peak intensity decreased (Fig. 1(d)). The maximum QY of the GQDs was 32.6%. After 120 min of reaction, a slight emission red-shift from 460 nm to 467 nm was observed. Spectrum changes were also observed in the PLE spectra as shown in Fig. 1(c). For the PLE spectra of the samples at longer than 120 min, the peak spectra changed significantly. Fig. 1(e) shows absorption spectra of the samples at various reaction times. After 120 min, the shape of the spectra changed drastically from one peak to two peaks. These results indicate that the molecular structure of the luminescent substance present at longer than 120 min is different from that of the samples present before 90 min.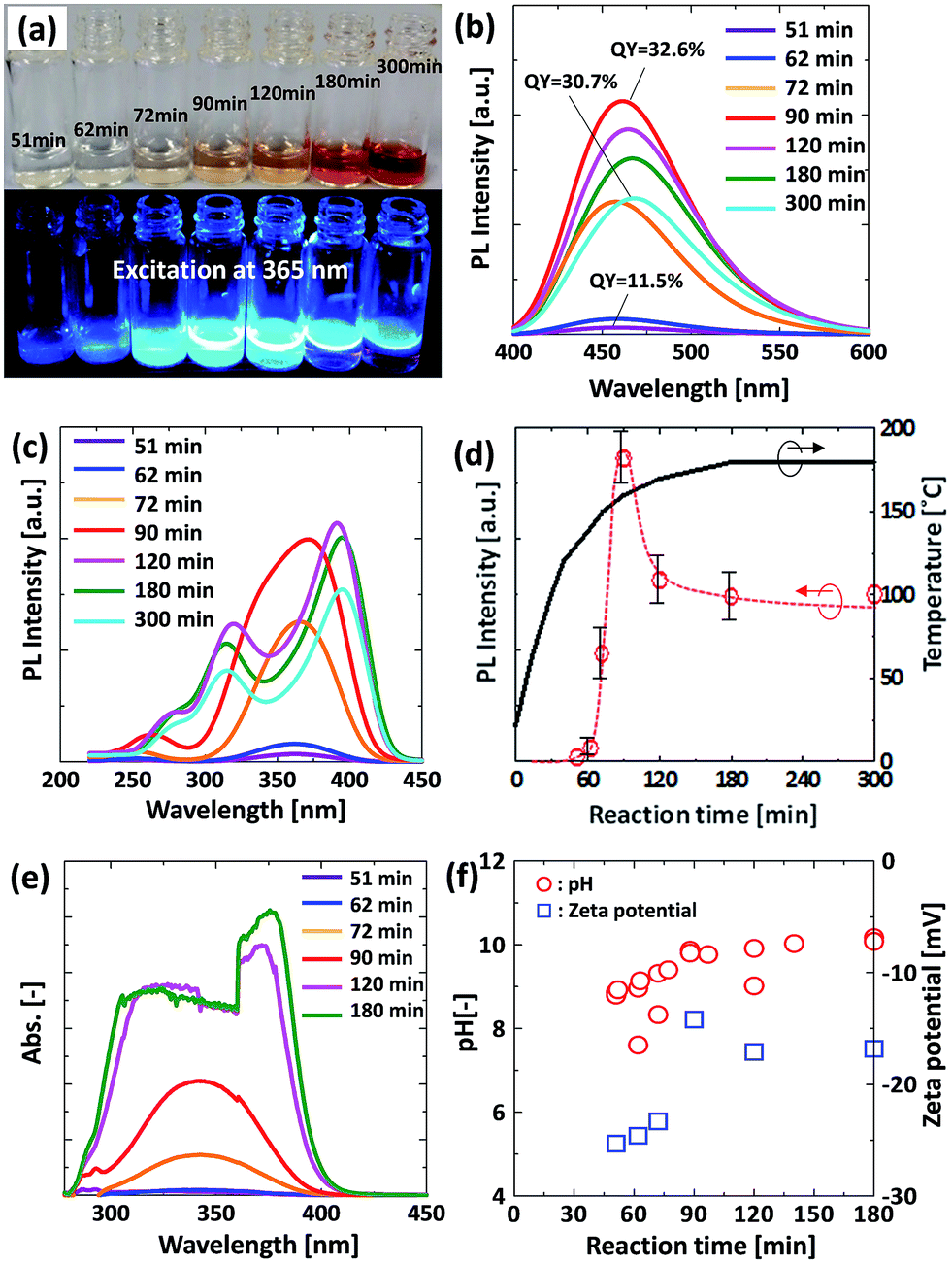 | ||
| Fig. 1 Effect of reaction time on the optical and physical properties of GQDs; (a) digital photograph, (b) PL spectra under excitation at 365 nm, (c) PLE spectra monitored at 460 nm, (d) PL intensity and temperature as a function of reaction time, (e) UV-vis absorption spectra, and (f) pH and zeta potential of solution samples as a function of reaction time. | ||
Fig. 1(f) shows pH and zeta potential changes as a function of reaction time. The pH of the solution changed from 7.6 to around 10.2. This change to a basic solution comes from ammonia generation because of urea decomposition during the reaction. According to previous literature, the zeta potential of carbon dots (C-dots) that were prepared by a microwave method from citric acid and urea was positive because of the presence of abundant nitrogen containing groups on the surface of the C-dots.9 The zeta potential values of all our samples were negative, which might indicate that the generated nitrogen containing group reacted with citric acid in the hydrothermal process as explained in the following section.
Fig. 2 shows XPS, TEM images, PL properties, and the photo-stability of the sample that were prepared at a reaction time of 90 min. The full scan XPS spectrum of the sample, as shown in Fig. 2(a), presents 3 peaks at 288, 399, and 532 eV, which corresponds to C1s, N1s, and O1s, respectively. This indicates that the sample is doped with nitrogen, which arose from urea. The high-resolution C1s spectrum demonstrates two peaks at 284.8, and 288.7 (Fig. 2(b)). The binding energy peak at 284.8 eV is ascribed to C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–H bonds, and the binding energy peak at 288.7 eV is attributed to OC–O, OC–N2 bonds, respectively. The high-resolution N1s spectrum of the sample (Fig. 2(c)) shows one peak at 399.6 eV, which are attributed to the pyrrolic N (C–N–C). The high-resolution O1s spectrum of the sample (Fig. 2(d)) detects one peak at 532.2 eV, which are attributed to the CO, C–O–C, C–OH. The above results indicate that the basic structure of the sample is the heterocyclic compound unit. This result is similar to the previous literature.28 Fig. 2(e) and (f) shows typical TEM images of the sample prepared by hydrothermal synthesis. The produced particle are well dispersed, partially crystallized, and range between 1–5 nm in diameter. The emission peaks of the sample at various excitation wavelengths did not shift and the maximum emission wavelength remained at 460 nm (Fig. 2(g)), which implies that both the size and the surface state of the sample remain uniform.19 This property prevents autofluorescence during their application. Furthermore, the GQDs prepared using our method have high photo-stability as shown in Fig. 2(h). In this research, the strongest fluorescence emission band was observed when exciting at 365 nm and a high fluorescence QY of 32.6% was obtained, which is 2.2 times higher than that of previously reported carbon-related luminescent materials, e.g. microwave-synthesized C-dots (14%) from citric acid and urea7 and GQDs prepared by pyrolyzing citric acid (9%).27
C, C–H bonds, and the binding energy peak at 288.7 eV is attributed to OC–O, OC–N2 bonds, respectively. The high-resolution N1s spectrum of the sample (Fig. 2(c)) shows one peak at 399.6 eV, which are attributed to the pyrrolic N (C–N–C). The high-resolution O1s spectrum of the sample (Fig. 2(d)) detects one peak at 532.2 eV, which are attributed to the CO, C–O–C, C–OH. The above results indicate that the basic structure of the sample is the heterocyclic compound unit. This result is similar to the previous literature.28 Fig. 2(e) and (f) shows typical TEM images of the sample prepared by hydrothermal synthesis. The produced particle are well dispersed, partially crystallized, and range between 1–5 nm in diameter. The emission peaks of the sample at various excitation wavelengths did not shift and the maximum emission wavelength remained at 460 nm (Fig. 2(g)), which implies that both the size and the surface state of the sample remain uniform.19 This property prevents autofluorescence during their application. Furthermore, the GQDs prepared using our method have high photo-stability as shown in Fig. 2(h). In this research, the strongest fluorescence emission band was observed when exciting at 365 nm and a high fluorescence QY of 32.6% was obtained, which is 2.2 times higher than that of previously reported carbon-related luminescent materials, e.g. microwave-synthesized C-dots (14%) from citric acid and urea7 and GQDs prepared by pyrolyzing citric acid (9%).27
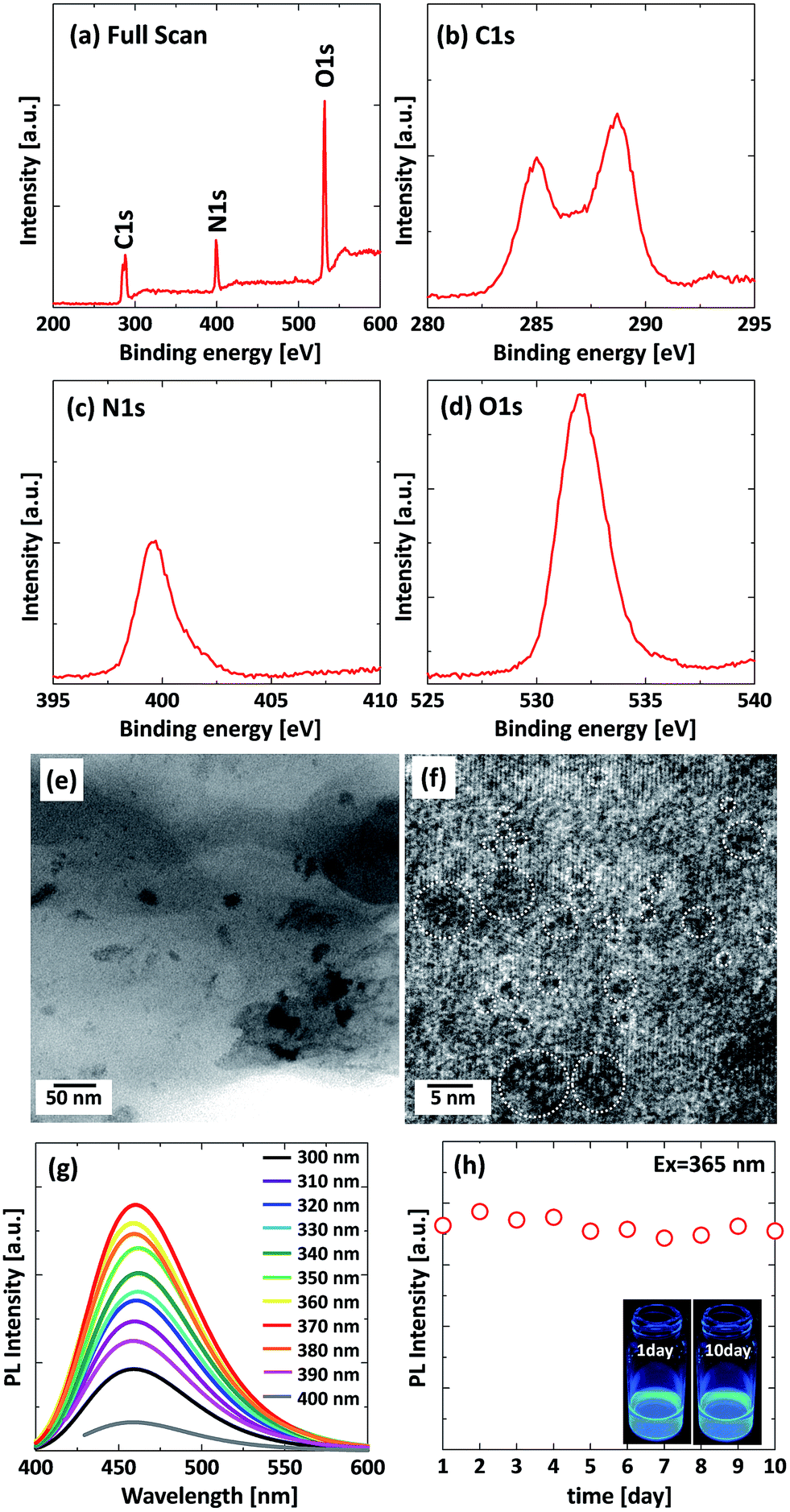 | ||
| Fig. 2 XPS, TEM and PL properties of the sample prepared at 90 min of reaction time; (a) XPS spectra, (b), (c) and (d) are the corresponding C1s spectrum, N1s spectrum and O1s spectrum. (e and f) TEM images (g) PL spectra of the sample at various excitation wavelengths from 300–400 nm, (h) stability of PL intensity of the sample, the inset is the digital photograph of sample, under 365 nm excitation at 1 day and 10 days. | ||
Based on the above-mentioned results, we found that the PL intensity of the GQDs has an optimum reaction time and excess reaction causes a quenching of the PL intensity because of significant absorption by the generated carbon materials. Therefore, the intermediate substance obtained before the formation of the GQDs has the potential to become a high QY luminescent material.
To investigate luminescent substances from citric acid, a NMR analysis was conducted for the samples prepared at reaction times of 51, 62, 72, 90, 120 and 180 min (Fig. 3). Fig. 3(a) shows a HSQC mapping image for the samples obtained at 51, 90, and 180 min. Fig. 3(b) shows an enlarged HSQC result. Fig. 3(c)–(f) shows 1D spectra for the lines from the origin (O), product 1 (P1), product 2 (P2), and product 3 (P3), as indicated in Fig. 3(a). From Fig. 3(c), the biggest peak was obtained at a reaction time of 51 min and this peak gradually decreased as the reaction time increased. The observed peak came from citric acid. Fig. 3(d) shows the intensity change for P1. The amount of product 1 gradually increased and reached a maximum value at 90 min, and this peak disappeared at a reaction time of 180 min. Fig. 3(f) shows the change in intensity for P3, which behaved in a similar manner to P1. Conversely, the maximum peak for P2 was obtained at 62 min and this peak gradually decreased and disappeared at 180 min (Fig. 3(e)). This peak indicates urea decomposition because the decomposition temperature of urea is around 150 °C. Based on these peak changes, in the first stage of the reaction, P2 formed and then P1 and P3 formed. Therefore, P1 and P3 are substances that contain different functional groups in the same molecule.
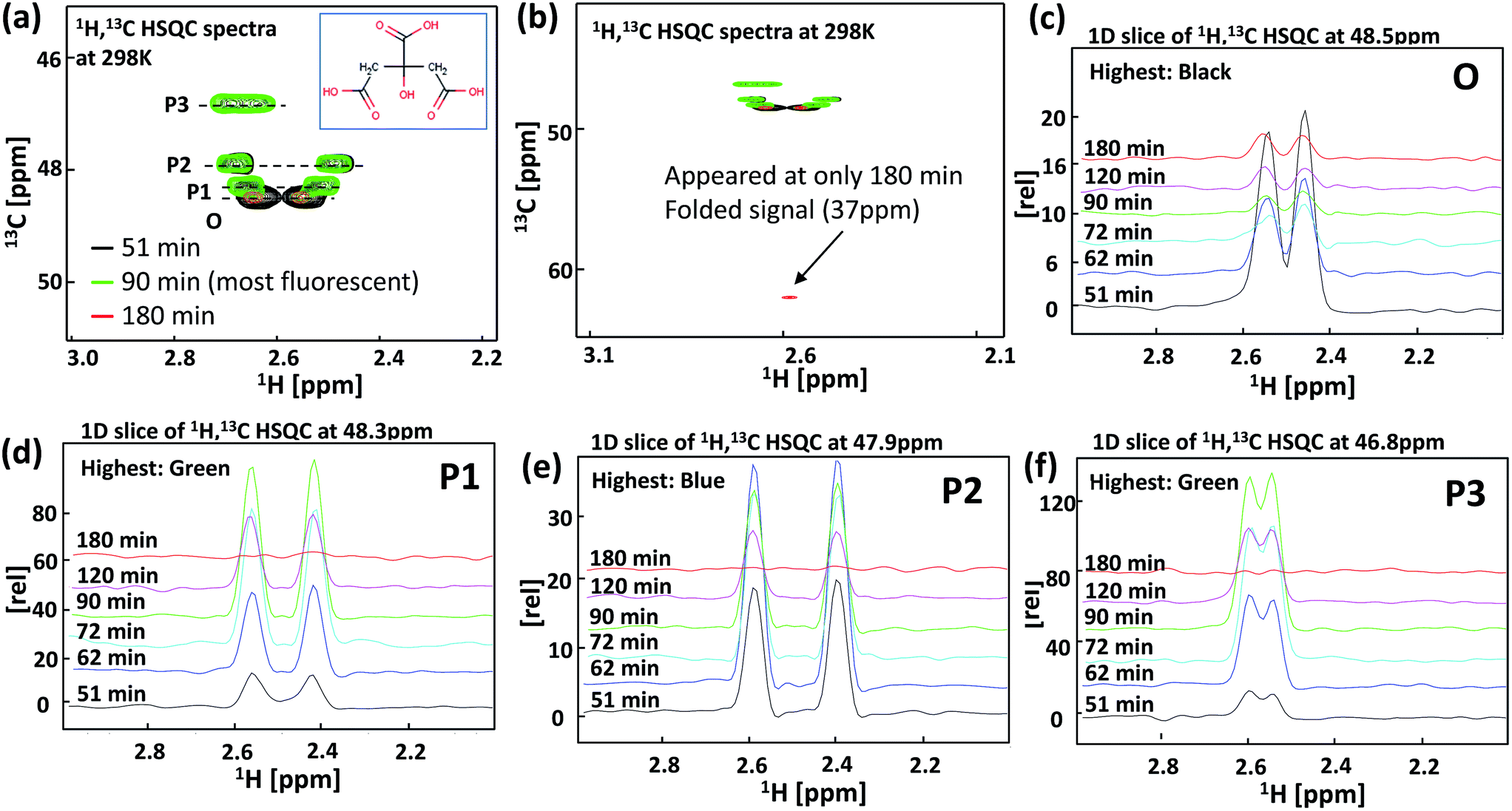 | ||
| Fig. 3 NMR analysis of the GQDs under the various reaction times; (a) HSQC mapping image (b) large area of HSQC, (c–f) 1D spectra on the lines of origin (O), product 1 (P1), product 2 (P2), and product 3 (P3) indicated in (a). | ||
According to the reaction process, citric acid and urea react first and P2 is formed. The molecule that arises from the signal of P1 and P3 is then formed via the hydrolysis of P2. Additionally, Fig. 3(b) shows an enlarged area HSQC result. The signal that appeared at 180 min indicates the decarboxylation of the product. Because the line shapes of the P1, P2 and P3 signals are almost the same as that of the O signal, the observed molecule is not very large and might be a dimer or oligomer of citric acid. However, further experiments and extensive effort such as high performance liquid chromatography (HPLS) analysis using reverse-phase column chromatography are required for an understanding of the molecular structures of the emission substances. Nonetheless, it is very clear from the experimental results that the molecular structure of citric acid is related to the highly luminescent substances.
Based on the NMR analyses, a possible formation mechanism of condensation product of citric acid amide via reaction of citric acid and urea in the hydrothermal synthesis is suggested in Fig. 4. In the initial stage of the reaction, citric acid amide is formed in a reaction between citric acid and urea. Further heating results in citric acid amide self-assembling into a nanosheet structure upon a dehydration and deammoniation between the carboxyl, hydroxyl, and amino groups of the intermolecular compounds under hydrothermal conditions. Because the monomers of citric acid amide have no emissions and the GQDs generated by the excess reaction have low emission, the strong luminescence is attributed to the dimers or oligomer-sized aggregates or the condensation product of citric acid amide.
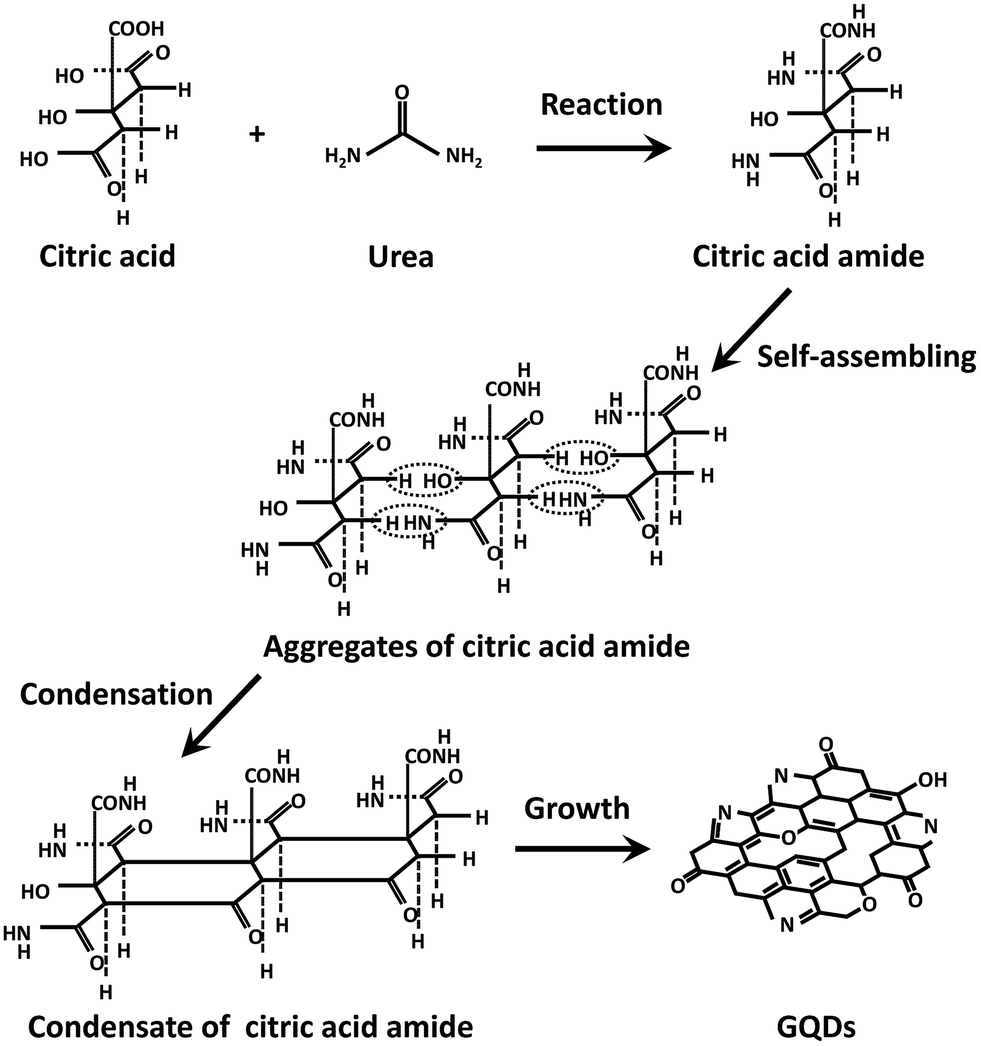 | ||
| Fig. 4 Schematic image of the formation of GQDs via reaction of citric acid and urea in the hydrothermal synthesis. | ||
According to previous literature, the emission mechanism of the GQDs depends on the ordered sp2 clusters with sizes of about 1 nm that are isolated within the sp3 C–O matrix. The presence of localized finite-sized molecular sp2 clusters within a sp3 matrix can lead to the confinement of π-electrons in GO. The radiative recombination of electron–hole pairs in these sp2 clusters can give rise to fluorescence.15,29 As mentioned above, the condensation product of citric acid amide contains similar structures and oxygen-containing functional groups and it is thus reasonable to assert that the origin of luminescence is the same as for the GQDs.
To future understand the luminescence mechanism, we focused on the type of precursor solution. According to previous reports, various carbon sources such as hexa-peri-hexabenzocoronene,24 graphene oxide,8 sodium citrate,27 glucose,25 and citric acid26 have been used for the preparation of GQDs. Among these carbon sources, citric acid derived GQDs exhibit a high QY. This result implies that citric acid related substances contribute to the high QY of the luminescent materials. In this experiment, the effect of various carbon sources, i.e. oxalic acid (OA), tartaric acid (TA), glutaric acid (GA), citric acid (CA), tetra ethylene glycol (TEG), and polyethylene glycol (PEG) on the PL properties was evaluated. To the best of our knowledge, the use of OA, TA, and GA as a carbon source for GQDs has not been reported to date. The physical properties of the carbon sources are shown in ESI (Fig. S3†). The PL and PLE spectra of the prepared samples are shown in Fig. 5. The experimental conditions for all samples were the same. Interestingly, a strong emission was observed only for the CA derived sample and weak emissions were observed for the other samples. This result supports the assertion that six-membered ring structure formation is essential for a high luminescence yield. CA supposedly forms six-membered ring structures by the aggregation of CA monomers in the hydrothermal reaction.26,30 Although GA has a similar structure to CA, it does not meet the requirements of aromatization because GA has no elimination group at the centre of its molecular structure. As a result, the GA derived sample has a weak PL intensity.
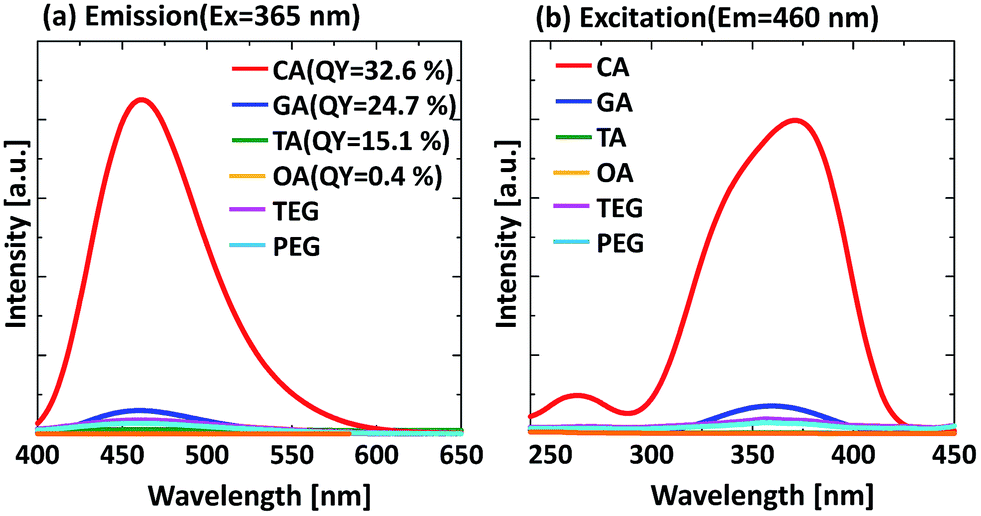 | ||
| Fig. 5 Effect of carbon source on the PL properties of samples prepared at 90 min of reaction time, (a) emission spectra, (b) excitation spectra; CA: citric acid, GA: glutaric acid, TA: tartaric acid, OA: oxalic acid, TEG: tetra ethylene glycol, PEG: polyethylene glycol. | ||
In previous research, Dong et al. synthesized the GQDs from citric acid only and the QY of the produced GQDs was 9.0%. For comparison, we synthesized the GQDs via hydrothermal synthesis using pure citric acid. From the PL and PLE result shown in Fig. 6, the PL intensity of the sample prepared from citric acid and urea is 40 times higher than that of pure citric acid. This result suggests that urea addition leads to a significant enhancement of PL intensity.
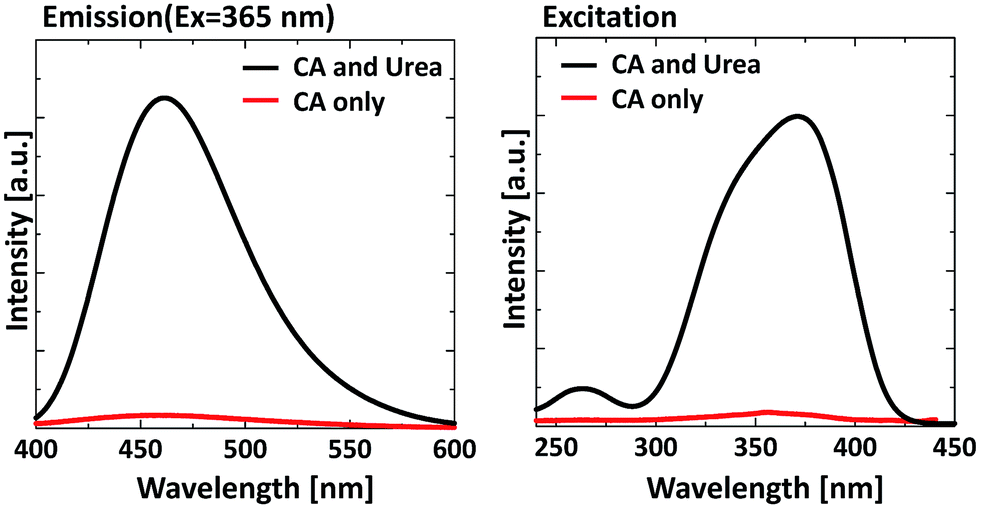 | ||
| Fig. 6 Effect of urea addition on the PL properties of GQDs, (a) emission spectrum, (b) excitation spectrum. | ||
The reason for this luminescence enhancement is the interaction between water and the COOH/CONH2 functional groups. In the case of citric acid only, since the interaction between water and COOH is strong, hydrogen bonding between the citric acid molecules becomes weaker. In contrast, when urea is added to the precursor solution, CONH2 bonding occurs as shown in Fig. 4. Therefore, hydrogen bonding between the citric acid amide molecules becomes stronger because the CONH2 function is not affected by water. As a result, the molecule is strongly-immobilized and the PL intensity increases.
Another reason is the partial nitrogen-doping effect. Some researchers have reported nitrogen doped GQDs using urea.26,28,30,31 These studies suggest that the nitrogen compounds in urea can react with the surface of carbon and act as a PL intensity enhancement agent during the formation of the GQDs. To confirm the existence of nitrogen bonding in our GQDs, the sample prepared in 90 min and with pure citric acid was analysed by FTIR (Fig. 7). The broad absorption bands present at 3000–3500 cm−1 are assigned to O–H and N–H stretching vibrations, and the bands at 1709 and 1667 cm−1 are attributed to CO in the COOH and CONH vibrational absorption bands, respectively. Compared with pure citric acid, the FT-IR spectrum of the prepared sample shows a peak shift from 1742 cm−1 and 1686 cm−1 to 1674 cm−1 and 1585 cm−1, which is the evidence for the existence of amide bonding in the products. Additionally, the absorption bands detected at around 3427 cm−1 and 3329 cm−1 are assigned to N–H and O–H. These functional groups improve the hydrophilicity and stability of the GQDs in aqueous systems.
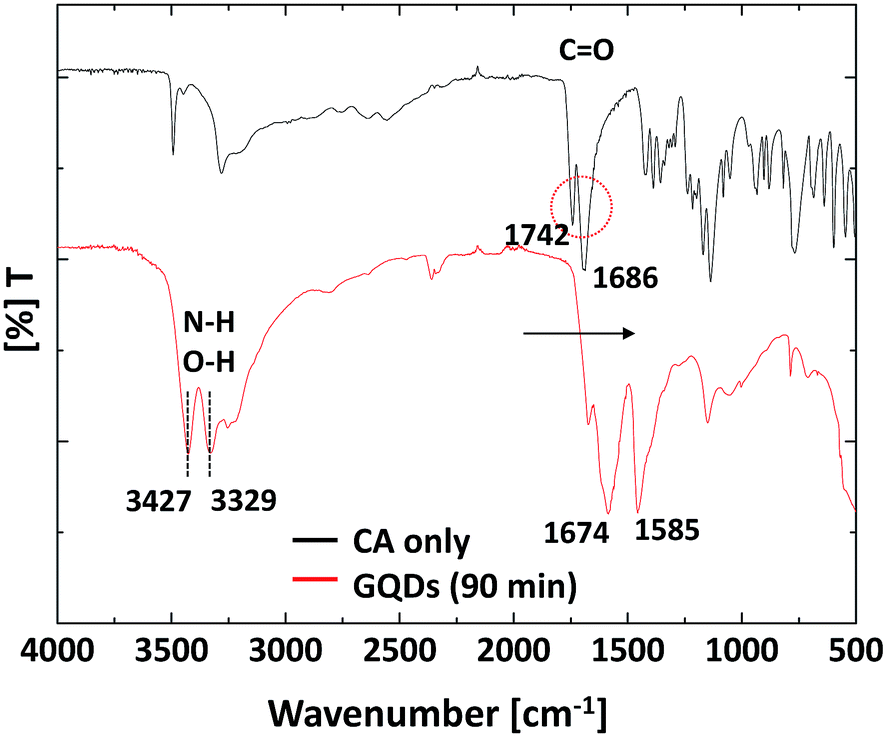 | ||
| Fig. 7 FT-IR spectra of the GQDs and CA (citric acid). | ||
Finally, we produced the GQDs–PVA composites by a simple mixing and drying process. Fig. 8 shows a digital photograph and PL spectra of the GQDs–PVA composites. The digital photographs show that the GQDs–PVA composite is transparent with good optical quality. The transmission spectra clearly reveal that the GQDs–PVA composite exhibits good transparency in the visible and near-IR region with a transmittance (T) of ca. 80% at wavelengths of 400–700 nm (Fig. 8(b)). From the PL properties (Fig. 8(c)), the strongest fluorescence emission was centred at 446.8 nm under 365 nm excitation with an enhanced QY of 43.6%. This result indicates that these aqueous solutions can be used as a new type of luminescent ink. Furthermore, this hybrid material is mechanically stable, can be easily cut, and it can be polished into different geometries and sizes depending on the target application and the detailed optical design.
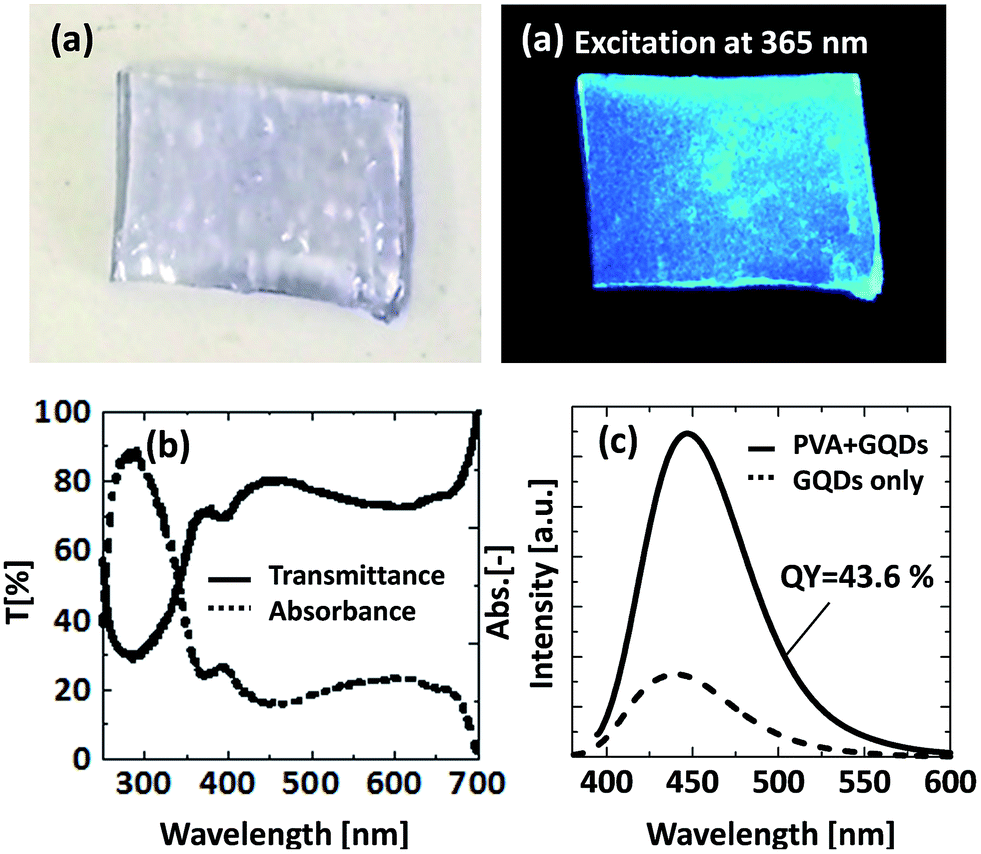 | ||
| Fig. 8 (a) Digital photographs, (b) UV-vis absorption, and (c) PL properties of the GQDs–PVA composite. | ||
Conclusions
We evaluated the transient nature of the PL properties and molecular structure on the GQDs formation via a hydrothermal reaction in detail. The results revealed the following interesting phenomena for the GQDs materials: (1) the GQDs have an optimum reaction time for a high QY; (2) NMR analysis revealed that the molecular structure that gave a high QY is an aggregate or a condensation product of citric acid amide; (3) the structure of citric acid is responsible for the significant luminescence of the GQDs; (4) urea addition to the citric acid solution leads to a significant enhancement in PL intensity, which is 40 times higher than obtained when using citric acid only. The as-prepared GQDs can be homogeneously mixed in PVA and this GQDs–PVA composite has high transparency and a high QY. We believe that these new GQDs findings open a new window for the preparation of excellent GQDs in less reaction time, and they provide an understanding of the luminescence mechanism of the GQDs.Acknowledgements
This work was supported by JSPS KAKENHI Grant numbers 26709061 and 24656413. We thank Dr Eishi Tanabe from the Hiroshima Prefectural Institute of Industrial Science and Technology for helping with TEM analyses, and Drs Naoya Tochio and Junichi Kakimura from Hiroshima University for NMR analyses.Notes and references
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736 CrossRef CAS PubMed
.
- S.-J. Yu, M.-W. Kang, H.-C. Chang, K.-M. Chen and Y.-C. Yu, J. Am. Chem. Soc., 2005, 127, 17604 CrossRef CAS PubMed
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726 CrossRef CAS PubMed
- T. Ogi, Y. Kaihatsu, F. Iskandar, W. N. Wang and K. Okuyama, Adv. Mater., 2008, 20, 3235 CrossRef CAS
- W.-N. Wang, T. Ogi, Y. Kaihatsu, F. Iskandar and K. Okuyama, J. Mater. Chem., 2011, 21, 5183 RSC
- T. Ogi, H. Iwasaki, A. B. D. Nandiyanto, F. Iskandar, W.-H. Wang and K. Okuyama, J. Mater. Chem. C, 2014, 2, 4297 RSC
- S. Qu, X. Wang, Q. Lu, X. Liu and L. Wang, Angew. Chem., Int. Ed., 2012, 124, 12381 CrossRef
- S. Zhu, J. Zhang, C. Qiao, S. Tang, Y. Li, W. Yuan, B. Li, L. Tian, F. Liu and R. Hu, Chem. Commun., 2011, 47, 6858 RSC
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S. T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430 CrossRef CAS PubMed
- Y. Liu and P. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 3362 CAS
- H. X. Zhao, L. Q. Liu, Z. De Liu, Y. Wang, X. J. Zhao and C. Z. Huang, Chem. Commun., 2011, 47, 2604 RSC
- F. Wang, Y.-h. Chen, C.-y. Liu and D.-g. Ma, Chem. Commun., 2011, 47, 3502 RSC
- X. Yan, X. Cui, B. Li and L.-s. Li, Nano Lett., 2010, 10, 1869 CrossRef CAS PubMed
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734 CrossRef CAS PubMed
- L. Li, G. Wu, G. Yang, J. Peng, J. Zhao and J.-J. Zhu, Nanoscale, 2013, 5, 4015 RSC
- X. Zhou, Y. Zhang, C. Wang, X. Wu, Y. Yang, B. Zheng, H. Wu, S. Guo and J. Zhang, ACS Nano, 2012, 6, 6592 CrossRef CAS PubMed
- J. Shen, Y. Zhu, C. Chen, X. Yang and C. Li, Chem. Commun., 2011, 47, 2580 RSC
- L. L. Li, J. Ji, R. Fei, C. Z. Wang, Q. Lu, J. R. Zhang, L. P. Jiang and J. J. Zhu, Adv. Funct. Mater., 2012, 22, 2971 CrossRef CAS
- S. Zhuo, M. Shao and S.-T. Lee, ACS Nano, 2012, 6, 1059 CrossRef CAS PubMed
- Y. Li, Y. Hu, Y. Zhao, G. Shi, L. Deng, Y. Hou and L. Qu, Adv. Mater., 2011, 23, 776 CrossRef CAS PubMed
- S.-S. Kim, J.-Y. Choi, K. Kim and B.-H. Sohn, Nanotechnology, 2012, 23, 125301 CrossRef PubMed
- X. Yan, X. Cui and L.-s. Li, J. Am. Chem. Soc., 2010, 132, 5944 CrossRef CAS PubMed
- L. Tang, R. Ji, X. Cao, J. Lin, H. Jiang, X. Li, K. S. Teng, C. M. Luk, S. Zeng and J. Hao, ACS Nano, 2012, 6, 5102 CrossRef CAS PubMed
- Y. Dong, J. Shao, C. Chen, H. Li, R. Wang, Y. Chi, X. Lin and G. Chen, Carbon, 2012, 50, 4738 CrossRef CAS PubMed
- Z. L. Wu, M. X. Gao, T. T. Wang, X. Y. Wan, L. L. Zheng and C. Z. Huang, Nanoscale, 2014, 6, 3868 RSC
- J. Lu, P. S. E. Yeo, C. K. Gan, P. Wu and K. P. Loh, Nat. Nanotechnol., 2011, 6, 247 CrossRef CAS PubMed
- R. Liu, D. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2011, 133, 15221 CrossRef CAS PubMed
- J. Zhou, Y. Yang and C.-y. Zhang, Chem. Commun., 2013, 49, 8605 RSC
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015 CrossRef CAS PubMed
- D. Qu, M. Zheng, P. Du, Y. Zhou, L. Zhang, D. Li, H. Tan, Z. Zhao, Z. Xie and Z. Sun, Nanoscale, 2013, 5, 12272 RSC
- D. Jiang, Y. Zhang, H. Chu, J. Liu, J. Wan and M. Chen, RSC Adv., 2014, 4, 16163 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09159k |
| This journal is © The Royal Society of Chemistry 2014 |