
Electrocatalytic degradation of 2,4-dichlorophenol using a Pd/graphene gas-diffusion electrode
Abstract
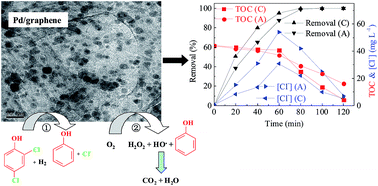
Palladium-modified graphene gas-diffusion cathodes were prepared using Pd/graphene catalysts and characterized using cyclic voltammetry, scanning electron microscopy, transmission electron microscopy, X-ray diffraction, Raman spectroscopy and Fourier transform infrared spectrometry. The Pd particles were amorphous and had an average size of 5.4 nm, and were highly dispersed in the graphene. A diaphragm electrolysis system sequentially fed with H2 and air over the gas-diffusion cathodes was constructed, and applied to the degradation of 2,4-dichlorophenol (2,4-DCP). When the Pd/graphene gas-diffusion cathode was fed with hydrogen, reductive dechlorination of 2,4-DCP took place, whereas acceleration of two-electron reduction of O2 to H2O2 proceeded in air. Dechlorination of 2,4-DCP reached approximately 96.4% after 60 min, while its removal efficiency and its removal in terms of total organic carbon (TOC) reached approximately 100% and 90.5%, respectively, after 120 min. By analysis of the electrolysis products by HPLC and IC, a reaction pathway has been proposed for the degradation of 2,4-DCP.
 Please wait while we load your content...
Please wait while we load your content...

