
Affibody conjugation onto bacterial cellulose tubes and bioseparation of human serum albumin†
Hannes Orelma*a,
Luis O. Moralesa,
Leena-Sisko Johanssona,
Ingrid C. Hoegerb,
Ilari Filpponena,
Cristina Castroc,
Orlando J. Rojasab and
Janne Lainea
aAalto University, School of Chemical Technology, Department of Forest Products Technology, FI-00076, Espoo, Finland. E-mail: hannes.orelma@aalto.fi
bNorth Carolina State University, Departments of Forest Biomaterials and Chemical and Biomolecular Engineering, Raleigh, NC 27695, USA
cUniversidad Pontificia Bolivariana, School of Engineering, Circular 1 # 70-01, Medellín, Colombia
First published on 3rd October 2014
Abstract
We attached anti-human serum albumin (anti-HSA) affibody ligands on bacterial cellulose (BC) by EDC–NHS-mediated covalent conjugation and physical adsorption and demonstrate their application for tubular biofiltration of blood proteins. The BC fibrils were first modified by carboxymethyl cellulose (CMC) by incorporation of CMC in the BC culture medium, producing in situ a CMC–BC tubular network that was used as biofilter. Alternatively, BC carboxylation was carried out by alkaline TEMPO–NaBr–NaClO oxidation. The BC and modified BC, grown in the form of tubes or flat films, were characterized by using scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), and conductometric titration. Anti-HSA affibody conjugation onto carboxylated cellulose thin film was verified from sensogram data obtained by surface plasmon resonance (SPR). The HSA specific binding capacity of the carboxylated cellulose conjugated with anti-HSA via EDC–NHS was approximately eight-fold larger when compared to the carboxylated cellulose surface carrying physically adsorbed anti-HSA (∼81 compared to 10 ng cm−2, respectively). Further proof of protein binding via anti-HSA affibody conjugated on tubules of CMC- and TEMPO-oxidized BC was obtained by fluorescence imaging. Specific binding of tagged HSA resulted in a linear increase of fluorescence intensity as a function of tagged HSA concentration in the contacting solution.
Introduction
Wood-based cellulose nanofibrils (CNFs) are a promising biomaterial that could significantly offset the use of oil-based materials in the future.1,2 The strong gel-formation ability3 of CNF could potentially be utilized in casting super strong translucent films for food packaging4 and diagnostics.5 Relevant to this discussion is our earlier report on bioactive CNF for specific human IgG binding.6 However, deployment of cellulose in shapes different than flat films can be conveniently facilitated if it is grown from bacteria, especially, if the application demands highly-pure and crystalline cellulose.Some bacteria genus, including Gluconacetobacter, Agrobacterium, Pseudomonas, Rhizobium, and Sarcina, have the capability to synthesize cellulose (bacterial cellulose, BC) in the presence of glucose, phosphate, and oxygen.7,8 BC has a ribbon-like shape and high crystallinity index.9,10 Due to the replication of bacteria, nanocellulose fibrils in the BC pellicle form a branched tangled structure, which has good mechanical properties even in the wet state.11,12 BC produced from Gluconacetobacter strains has been used as a food supplement, in electronics and in several medical applications. Moreover, since the BC growth takes place only in the presence of oxygen, their assembly can be directed to form different shapes, depending on the air–water interface used, for example in closed vessels, tubes (BC-tubes), sheets, sacks, cylindrical balloons, etc.13–15 The non-toxicity and high stability of BC makes it an ideal material in medical applications such as artificial blood vessels16,17 and skin wound healing materials.18
Synthesized BC tubes have shown the potential to significantly resist the internal pressure,15 which is a requirement in biofiltration. It has also been shown that small molecules (molecular mass of the order of 20 kDa) can diffuse through BC pellicles. In addition, immunological cells like globulins (ca. 66 kDa) can be filtered out from solution.19,20 Therefore, BC tubes could potentially be utilized in biofiltration assays, such as in separation of immunological proteins like antibodies (ca. 150 kDa). Moreover, the incorporation of antibodies, peptide ligands, protein A, etc. onto the inner walls of BC tubes21,22 opens new possibilities in biofiltration for detection and separation of specific target proteins.
Several methods for introducing functional groups on BC fibrils have been reported, including in situ and ex situ methods. In the case of ex situ modifications, carboxylation via TEMPO-oxidation23 and amination using diethylenetriamine24 are amongst the most utilized modification chemistries. In addition, most of the modification methods reported for cellulose can also be exploited with BC.25 A simple and elegant in situ method is to add cellulosic derivatives into the culture medium. Such system, i.e., cellulose fibrils encapsulated with cellulosic derivatives during the incubation process, resembles that of heteropolysaccharides in contact with plant cell walls. Cellulose derivatives such as xyloglucan,26 carboxymethyl cellulose (CMC),27,28 chitosan,29 and hydroxylethylcellulose30 have been successfully utilized for the in situ modification of BC. Furthermore, some water soluble polymers, such as polyethylene oxide31 and polyvinyl alcohol (PVA)32 have been used to in situ functionalize BC.
In this communication the concept of in situ modification of BC tubes with CMC (CMC–BC tubes) for selective biofiltration is demonstrated. Affibodies, i.e., engineered proteins that mimic the antigen binding regions of native antibodies, were used as active molecules mainly because of their wide availability. They have similar sensitivity and affinity properties when compared to native antibodies.33 Moreover, since affibodies are produced by modified bacteria and not in living eukaryotes, they can be modified for specific protein detection, which in turn opens new venues for practical deployment. CMC–BC tubes were obtained from Gluconacetobacter medellinensis grown in a culture medium in the presence of dissolved CMC. The selection of CMC was based on its non-toxicity for human cells,34 availability, and suitability for anchoring antibodies onto cellulose.35 As an alternative carboxylation reference, an alkaline TEMPO–NaBr–NaClO oxidation of BC was performed and tested. BC tubes with and without CMC were characterized by conductometric titration, scanning electron microscopy (SEM), and X-ray photoelectron microscopy (XPS). Chemical reactions on cellulose were demonstrated using surface plasmon resonance (SPR) on thin cellulose films prepared by Langmuir–Schaeffer method. The specific protein detection of synthetized BC tubes was verified with fluorescence-stained human serum albumin (HSA). Scheme 1 offers an illustration of the developed materials and concepts noting that affibody modification makes them generic for use in the detection and separation of diverse antigens and plasma proteins.
 | ||
| Scheme 1 Schematic illustration of the synthesis of CMC-modified bacterial cellulose tubes (BC tubes) and their subsequent functionalization with affibodies for biofiltration. | ||
Experimental section
Gluconacetobacter medellinensis (G. medellinensis) was provided by the School of Engineering, Universidad Pontificia Bolivariana, Colombia and the properties of the strain are described elsewhere.36 CMC (DS of 0.7, Mw of 250 kDa, #419311), D-(+)-glucose (#G5767), yeast extract (#Y1625), sodium phosphate dibasic (Na2HPO4, #S3264), bacteriological peptone (#0556), NHS (N-hydroxysuccinimide, #130672), EDC (1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride, #03450), HSA (#A9511), and TEMPO free radical ((2,2,6,6-tetramethyl-piperidin-1-yl)oxyl, #426369) were obtained from Sigma-Aldrich (Helsinki, Finland). Anti-HSA (anti-human serum albumin) affibody dimer molecules (#ab31897, Mw 14 kDa) were obtained from Abcam plc. (Cambridge, UK), and used following the manufacturer instructions. The HSA binding affinity of anti-HSA affibody is kd = 10 nM (value obtained from Affibody AB, Sweden). All chemicals were used without any purification steps. All other chemicals used in this study were analytical grade and used without any purification steps. The water used in all experiments was deionized and further purified with a Millipore Synergy UV unit (MilliQ-water).Synthesis of CMC-modified BC membranes and tubes
BC was synthesized from G. medellinensis36 in a standard Hestrin–Schramm (HS) medium.37 The culture medium was prepared by dissolving 20 g glucose, 5 g yeast extract, 5 g bacterial peptone, and 2.5 g Na2HPO4 in a 1 L of deionized water, and the pH was adjusted to 4 with citric acid. CMC (0–5 g L−1) was dissolved in the culture medium, and the medium was sterilized with an autoclave (120 °C for 20 min). G. medellinensis was statically incubated at 28 °C for 9 days. For the charge and water retention capacity (WRV) measurements, BC was synthetized as a sheet in a shallow container with a large air–water interface, in order to ensure high volumes of BC. For XPS, SEM, and filtration tests BC was incubated in a closed vessel, where the air inlet takes place through a silicone tube, which allows BC to grow only around the tube. The diameter and wall thickness of the silicone tubes were 10 and 1 mm, respectively. BC tubes were collected after incubation. The CMC concentration used for incubating BC tubes was 2 g L−1. After incubation, BC sheets and tubes were purified by boiling first in 0.1 M NaOH at 60 °C for 4 hours and then in MilliQ-purified water at 60 °C for 1 hour as described elsewhere.15 Thereafter, synthetized BC was rinsed several times with MilliQ-purified water at room temperature to remove bacteria residues and loosely bound CMC. Finally all samples were sterilized by boiling in MilliQ-water for couple minutes. All samples were stored in sterilized laboratory bottles at 4 °C and used within one week. The length of a synthesized BC tube was approximately 20 cm and the wall thickness 1.8 ± 0.2 mm in the wet state.TEMPO-oxidation of unmodified BC sheets and tubes
The alkaline TEMPO-oxidation system was employed as an alternative route for the carboxylation of bacterial cellulose.23 Unmodified BC sheets and tubes were TEMPO-oxidized by using the 2,2,6,6,-tetramethylpipelidine-1-oxyl radical (TEMPO)–NaBr–NaClO system that consisted of 0.13 mmol TEMPO and 4.7 mmol NaBr dissolved in 100 mL water to which 5.65 mmol NaOCl was added and the solution pH adjusted to 10 by 1 M HCl. BC sheets or tubes (sheet size of approximately 5 × 5 cm2) placed in the prepared TEMPO solution, and the pH of the system was fixed to 10 by 1 M NaOH addition. Samples were kept in the TEMPO solution from 1 to 30 min, and the oxidation reaction was quenched by immersing the samples in 50 w% ethanol–water for 5 min followed by washing with a large amount of MilliQ-purified water for several hours to remove the excess of carboxylation chemicals. Finally, the TEMPO-oxidized samples were boiled in MilliQ-purified water and stored at 4 °C as the CMC–BC-samples.Charge density and water retention value of modified BC
The amount of weak acid groups (carboxyls) of CMC-modified and TEMPO-oxidized BC was determined by a conductometric titrator 751 GPD Titrino (Metrohm AG, Herisau, Switzerland) following a standard SCAN-CM 65:02. The analyses were carried out with BC sheets (size approximately 10 × 10 cm2), because the dry mass required for the accurate carboxyl content determination is higher that can be obtained from the BC tubes. Samples were first washed with 0.01 M HCl for 1 h and then disintegrated in water with a tip ultrasonificator (40% amplitude for 10 min). The charge determination by the conductometric titrations were performed by 0.025 mL injections of 0.1 M NaOH using 30 s intervals. The amount of weak acid (carboxyl) groups was calculated as described in the standard method (SCAN-CM 65:02).The effects of the added CMC or TEMPO-oxidation on the volume of bound water in BC were determined following the standard method SCAN-C 62:00. All analyses were performed with both never-dried and air-dried BC samples in order to estimate the porosity changes and degree of irreversible cellulose collapse (hornification). All measurements were repeated at least four times using BC sheets (size 4 × 4 cm2).
Surface analysis of modified BC tubes by XPS
The surface chemical composition of BC tubes was examined using a Kratos Analytical AXIS Ultra electron spectrometer with a monochromatic Al Kα X-ray source at 100 W and a neutralizer. The XPS experiments were performed on the dry films, which were pre-evacuated overnight. At least three different spots of each sample were scanned. Spectra were collected at an electron take-off angle of 90° from sample areas less than one mm in diameter. Elemental surface compositions were determined from low-resolution measurements (160 eV pass energy and 1 eV step) while the surface chemistry was probed with high resolution measurements (20 eV pass energy and 0.1 eV step). The carbon C 1s high-resolution spectra were curve fitted using parameters defined for cellulosic materials38 and all binding energies were referenced to the aliphatic carbon component of the C 1s signal at 285.0 eV.39 According to the XPS reference used in situ (100% cellulose ash free filter paper measured along with each sample batch) the conditions in UHV remained satisfactory during the XPS experiments.38Imaging via SEM
The morphology of the CMC-modified and TEMPO-oxidized BC tubes was imaged by using a Jeol JSM 5910 LV microscope operated at 20 kV. The imaging was carried out on freeze-dried samples previously frozen using liquid nitrogen to preserve the structure of BC tubes. The samples were coated with gold/palladium using an ion sputter coater. Both cross-sections and inner surfaces of BC-tubes were imaged.Conjugation of anti-HSA affibodies onto BC tubes
The anti-HSA affibody molecules were covalently conjugated onto the CMC-modified (2 g L−1 CMC in the culture medium) and TEMPO-oxidized (TEMPO-oxidation for 10 min) BC-tubes by using EDC–NHS-mediated conjugation.40 First, tubes (approx. 3 cm long) were rinsed with 10 mM NaOAc buffer at pH 5 and then immersed in the activation solution (0.1 M EDC and 0.4 M NHS in 10 mM NaOAc buffer at pH 5) for 30 min. Next, the activated tubes were rinsed extensively with 10 mM NaOAc buffer in order to remove residual, spent activation chemicals, and then they were immersed in the anti-HSA affibody solution (0.1 mg mL−1 anti-HSA in 10 mM NaOAc at pH 5) for 60 min. After conjugation, the tubes were extensively rinsed first with 10 mM NaOAc at pH 5 and then with 10 mM phosphate buffer at pH 7.4 in order to remove any unconjugated anti-HSA. In the following step, the tubes were washed with 0.1 M ethanol amine at pH 8.5 for 30 min to remove unreacted NHS-esters, which can reduce the specificity of the prepared biointerface. Finally, the anti-HSA functionalized BC tubes were extensively rinsed with phosphate buffer at pH 7.4, and then immediately used for the HSA detection.HSA-detection with the anti-HSA functionalized BC tubes
The specific detection of HSA with anti-HSA functionalized BC tubes was carried out using fluorescence-stained HSA. HSA was labelled with dansylchloride (fluorescence dye) as described elsewhere.41 Dansylated–HSA was sequentially purified, first with a 10–12 kDa mesh dialyzation membrane tube (SpectraPor, Spectrumlabs) against MilliQ-purified water and then by washing six times with Amicon Ultra centrifugal filter tubes (Mw 30 kD) against 10 mM phosphate buffer at pH 7.4. Finally, the dansylated–HSA was freeze-dried and stored in a desiccator.Anti-HSA functionalized BC tubes (both CMC-modified and TEMPO-oxidized, length 1 cm) were added into the solution of dansylated–HSA (concentration varied from 0.001 to 0.1 mg mL−1, 10 mM phosphate buffer at pH 7.4) for 30 min and then the tubes were rinsed with 10 mM phosphate buffer at pH 7.4. The amount of dansylated–HSA bound onto the inner surface of anti-HSA functionalized BC tubes was analyzed by a Leica TCS SP2 confocal laser scanning microscope (Leica microsystems CMS GmbH, Manheim, Germany) with an excitation and detection wavelengths of 488 and 500–530 nm, respectively. The laser power was fixed to a constant and the image size of 750 × 750 μm2 was used in all CLSM measurements. Before the measurements, the samples were freeze-dried using liquid nitrogen.
Conjugation of anti-HSA onto CMC-modified cellulose thin films as determined by surface plasmon resonance (SPR)
The conjugation of anti-HSA on CMC-modified cellulose and the subsequent binding of HSA on the prepared anti-HSA–CMC biointerface were examined by using a SPR instrument (Model Navi 200, Oy BioNavis Ltd, Tampere, Finland). The thicknesses of the adsorbed layers were calculated by using eqn (1):42
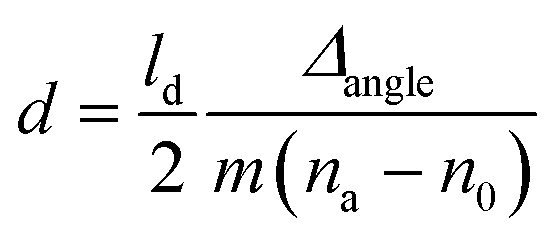 | (1) |
Langmuir–Schaefer cellulose films were deposited on gold wafers by using the deposition technique as described by Tammelin et al.46 The crystallinity, thickness, and roughness of similar films are 54%, 18 nm, and 0.5 nm, respectively.47 The gold wafers were first cleaned by using UV/ozone treatment followed by a spin coating of 0.1 w% polystyrene in toluene (4000 rpm, 30 s). The obtained polystyrene-coated wafers were then heat-treated in an oven at 60 °C for 10 min to ensure an uniform hydrophobic layer suitable for trimethylsilyl cellulose (TMSC) deposition. TMSC coated on SPR-wafers was converted to cellulose via desilylation with hydrochloric acid vapor as described elsewhere.48 Before the SPR experiments, the cellulose films were allowed to stabilize overnight in the respective buffer solution.
The conjugation of anti-HSA and subsequent detection of HSA were monitored by SPR as follows: first 0.5 g L−1 CMC in 25 mM NaCl at pH 5 was allowed to adsorb on a cellulose film in the SPR and then loosely bound CMC was rinsed out with 25 mM NaCl at pH 5. CMC-modified cellulose was activated with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 0.1 M EDC and 0.4 M NHS in 10 mM NaOAc at pH 5. Next, 0.1 g L−1 anti-HSA in 10 mM NaOAC at pH 5 was allowed to bind on the EDC–NHS activated CMC–cellulose surface. The surface was rinsed with 10 mM NaOAC at pH 5, 10 mM phosphate buffer at pH 7.4, 0.1 M ethanol amine at pH 8.5, and 10 mM phosphate buffer at pH 7.4, respectively. Then 0.1 g L−1 HSA in 10 mM phosphate buffer at pH 7.4 was allowed to adsorb on the prepared anti-HSA biointerface and SPR monitoring was terminated with rinsing 10 mM phosphate buffer at pH 7.4. As references, 0.1 g L−1 anti-HSA in 10 mM NaOAC at pH 5 was adsorbed on a CMC–cellulose surface without the EDC–NHS activation, and 0.1 g L−1 HSA in 10 mM phosphate buffer at pH 7.4 was adsorbed on both pure cellulose and CMC-modified cellulose.
:1 mixture of 0.1 M EDC and 0.4 M NHS in 10 mM NaOAc at pH 5. Next, 0.1 g L−1 anti-HSA in 10 mM NaOAC at pH 5 was allowed to bind on the EDC–NHS activated CMC–cellulose surface. The surface was rinsed with 10 mM NaOAC at pH 5, 10 mM phosphate buffer at pH 7.4, 0.1 M ethanol amine at pH 8.5, and 10 mM phosphate buffer at pH 7.4, respectively. Then 0.1 g L−1 HSA in 10 mM phosphate buffer at pH 7.4 was allowed to adsorb on the prepared anti-HSA biointerface and SPR monitoring was terminated with rinsing 10 mM phosphate buffer at pH 7.4. As references, 0.1 g L−1 anti-HSA in 10 mM NaOAC at pH 5 was adsorbed on a CMC–cellulose surface without the EDC–NHS activation, and 0.1 g L−1 HSA in 10 mM phosphate buffer at pH 7.4 was adsorbed on both pure cellulose and CMC-modified cellulose.
Results and discussion
Synthesis and structural characterization of CMC-modified BC tubes
The synthesis of CMC–BC tubes was carried out by using a closed incubation vessel equipped with a supporting silicon tube with constant oxygen flow for bacteria feeding (see Scheme 1). Synthetized CMC–BC tubes were uniform and without visible defects (Fig. 1a–c). The presence of CMC in the culture medium increased the wall thickness of BC tubes compared to that produced in its absence (wet wall thicknesses of 0.9 ± 0.1 and 1.8 ± 0.2 mm for BC and CMC–BC tubes, respectively). As a reference, purified BC tubes were TEMPO-oxidized for 10 min and no changes in thickness were observed (1.0 ± 0.1 mm). It should be mentioned that the use of CMC in a culture medium of Gluconacetobacter has been reported to alter the mechanism by which the bacteria assemble into BC fibrils.27 It was postulated that CMC disrupts the formation of fibrillar bundles (BC fibrils) by incorporating CMC into BC fibrils leaving the crystallization of microfibrils intact. Therefore, BC fibrils that are synthesized in the presence of CMC are more meandering and loosely packed than unmodified BC fibrils. It has also been reported that incorporated CMC accelerates the rate of cellulose synthesis by 30%.49 Hence, it is conceivable that the relatively high thickness of CMC–BC tubes originates from the loose packing of fibrils and the high biosynthesis rates. | ||
| Fig. 1 Photo images of the synthetized CMC-modified BC tubes before (a) and after (b) purification. The cross section images of synthetized BC tubes after different treatments (unmodified, 10 min TEMPO-oxidation, and 2 g L−1 CMC added in the culture medium) are also included (c). | ||
The morphology of the synthesized CMC–BC tubes were explored by SEM. Both, the inner surface and cross-section of CMC–BC tubes were analyzed. Fig. 2 clearly illustrates that the incorporation of CMC changes the morphology of BC. Fibrils of CMC–BC were more meandered and a slightly thicker when compared to unmodified BC fibrils (Fig. 2a and b). These effects are most probably due to the incorporation of CMC into BC fibrils that leads to a looser packing of microfibrils.27 Based on SEM images, the inner surface of CMC–BC tubes also contained more voids and pores than unmodified BC tubes (Fig. 3a and b), but the cross-sections were similar (Fig. 3c and d). The porosity of synthesized CMC–BC tubes was not characterized, but it has been reported that a high CMC concentration can slightly increase the pore size of BC.50 Thus, in the present case it is expected that the permeability of proteins through the BC is not be significantly altered by the small concentration of CMC used in the culture medium.
 | ||
| Fig. 2 Plane view SEM images of the inner-surfaces of BC (a) and CMC–BC (b) tubes. The magnification used in the images is 50000×. | ||
 | ||
| Fig. 3 Plane view SEM images of the inner-surfaces of BC (a) and CMC–BC (b) tubes. The SEM images of the corresponding cross-sections are also included in (c) and (d), respectively. The magnification used in the images is 5000×. | ||
The effect of CMC incorporation in BC charge and water retention values (WRV) was examined by using the corresponding BC pellicles. It is important to note here that the conductometric titration measures only the weak acid groups (carboxyls), and therefore the measured charge represents the total carboxyl content of the sample. As expected, the charge of unmodified BC was zero (Fig. 4a) since BC is pure, native cellulose and does not contain any weak acid groups. However, a low amount of CMC (0.1 g L−1) in the culture medium raised the charge of CMC–BC to 72 μeq. g−1. A charge plateau level was reached for CMC concentrations in the culture medium over 2 g L−1 (charge of ∼400 μeq. g−1). The highest concentration of CMC used was 5 g L−1, which was limited by the resultant viscosity of the solution (this in turn, may influence the formation of BC pellicles). The WRV measurements were performed to demonstrate the effect of CMC on the swelling of fibrillar networks and their water uptake capacity. The high WRV value (990%) of unmodified BC (Fig. 4a) was comparable to those previously reported in the literature.17 The addition of CMC in the culture medium increased the WRV of never-dried CMC–BC, and this increase was clearly associated with the electrostatic charge. The highest WRV values were obtained for samples containing over 2 g L−1 of CMC, which corresponds to the higher carboxyl content of CMC–BC. The WRV values were determined also for re-swollen dried CMC–BC pellicles, in order to monitor the irreversible structural changes (hornification) after drying. As expected, the WRV value for re-swollen, unmodified BC was significantly lower (240%) than that of never-dried BC (990%). This indicates that the drying leads to a partly collapsed fibrillar structure as a result of the hornification phenomenon. It was expected that CMC improves the irreversibility of structural changes in a BC-pellicle due to increased anionic charge. Interestingly, a CMC addition as low as 0.1 g L−1 raised the WRV value of swollen BC to levels as high as 400%, whereas the highest WRV value (530%) was observed after an addition of 5 g L−1. This indicates that the incorporation of CMC increases the swelling capacity of BC and provides resistance to irreversible structural changes.
 | ||
| Fig. 4 Water retention values (WRV) of never dried (circle symbols) and air-dried BC (triangle symbols) as a function of CMC added in the culture medium (a). The content of the carboxyl groups in BC as a function of CMC added in the culture medium is also shown (data corresponding to the right y-axis, square symbols). WRV values of never dried and air dried BC as a function of the TEMPO-oxidation time are shown in (b). The content of the carboxyl groups in BC as a function of the TEMPO-oxidation time is included (data corresponding to the right y-axis, square symbols). | ||
The alkaline TEMPO-oxidation was used as a reference method for the in situ CMC modification and to explore the topological effects related to the presence of carboxyl groups on the BC fibrils. The in situ CMC-modification is likely to produce BC fibrils with evenly distributed carboxyl groups, whereas the TEMPO-oxidation introduces carboxyls only on the surface of highly crystalline BC fibrils.23 The charge and WRV results (both never-dried and air-dried with re-swelling) of TEMPO-oxidized BC pellicles are presented in Fig. 4b. The results indicate that even a short TEMPO-oxidation (1 min) is enough to raise the charge of BC to a plateau level (charge about 220 μeq. g−1), after which the amount of carboxyl groups remains practically unchanged. This is not totally surprising as it is known that TEMPO-treatment oxidizes the easily accessible surface C6-hydroxyls and disordered regions within the first minutes of oxidation.51 However, regions with lower accessibility (higher crystallinity) require a longer oxidation time. For example, charges up to 800 μeq. g−1 have been reported for TEMPO-oxidized BC while oxidation times longer than 5 h were employed.52,53 Yet, it has been shown that the elevated TEMPO-oxidation significantly reduces the DP of cellulose microfibrils because of the depolymerization reactions.51,54 Therefore, short TEMPO-oxidation times are preferred when the strength properties of BC-fibrils are to be preserved. WRV of never-dried TEMPO-oxidized BC increased as a function of the oxidation time, while at the same time the charge remained almost constant (Fig. 4b). This indicates that TEMPO-oxidation increases the penetration of water in the BC network. Likely explanations are the increased polarity of oxidized BC-fibrils and more open joints between the individual fibrils. In general, the WRV of air-dried TEMPO-oxidized BC samples were considerable lower when compared to those of air-dried CMC–BC samples. This indicates that hornification occurs to a greater extent when the carboxyl groups are located only on the surface of BC-fibrils. However, it should be noted that after 30 min TEMPO-oxidation the WRV of the re-swollen sample was similar to that of the CMC-modified sample. This is most probably due to the aforementioned depolymerization reactions, which lower the DP of BC-fibrils and therefore make them more prone to swelling.54
The surface chemical composition of CMC–BC tubes was investigated by XPS. This method has been effectively used for exploring the chemistry of the outermost layers of cellulose.38 No significant differences between the XPS spectra of unmodified BC and a cellulose standard were found (Fig. 5). However, the O/C ratios of BC samples were slightly lower than that of the cellulose standard. The results indicate the presence of small amounts of impurities, possibly from the incubation process. In fact, the nitrogen signal (Table 1), which is not present in the pure cellulose standard, appears in BC samples. This is a clear indication that a small amount of residual protein (from bacteria) remain on the surfaces of BC fibrils. This small amount of protein can also contribute to the small unbalance in O/C ratio. Interestingly, the nitrogen signal was lower in the CMC–BC sample, which suggests that the incorporation of CMC in the culture medium may facilitate the separation of residual proteins during washing. However, the nitrogen signal observed for TEMPO-oxidized BC (TEMPO-oxidized after purification of BC) was almost as intense as that of unmodified BC, indicating that bacteria separation was not affected by charge-driven electrostatic repulsion to the same extent compared to the case of CMC–BC. Therefore, the hydrogel-like nature of CMC seems to be a dominant factor determining the diffusion of protein residues out from the surface of BC fibrils. The carboxylation of cellulose could not be revealed by XPS measurements (as reported by us and other groups),5,55 which is most probably due to the medium-dependent surface reconstruction of cellulose in dry media.56 However, a small sodium signal observed for CMC-modified and TEMPO-oxidized BC may be taken as indirect indication of the introduction of carboxyl groups onto BC fibrils (the carboxyl groups in CMC and TEMPO-oxidized cellulose exist as sodium carboxylates).
 | ||
| Fig. 5 XPS spectra of unmodified BC, CMC-modified BC (CMC–BC, 2 g L−1 CMC added in the culture medium), and TEMPO-oxidized BC (10 min TEMPO-oxidation). The spectra are shifted vertically to facilitate comparisons. | ||
| Sample | Element (at%) | C 1s component (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| O 1s | C 1s | N 1s | Na 1s | C(C–C) | C(C–O) | C(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) O) |
C(COO) | O/C | |
| Pure BC | 36.0 | 62.3 | 1.8 | nd. | 12.2 | 67.0 | 18.8 | 2.0 | 0.58 |
| BC with added CMC | 36.6 | 62.5 | 0.7 | 0.2 | 17.8 | 62.5 | 17.5 | 2.3 | 0.55 |
| BC with 10 min TEMPO-oxidation | 35.0 | 62.8 | 1.8 | 0.4 | 22.8 | 58.1 | 16.9 | 2.3 | 0.56 |
| Cellulose standard | 39.2 | 60.8 | nd. | nd. | 5.0 | 74.5 | 19.1 | 1.4 | 0.64 |
Conjugation of affibodies onto CMC-modified cellulose monitored by SPR
Cellulose thin films were utilized to verify the conjugation by using the surface plasmon resonance technique, SPR. First, cellulose films supported on gold sensors were modified by adsorbing CMC from electrolyte solution. As expected, CMC was found to adsorb irreversibly onto cellulose (Fig. 6a).57 It has been postulated that the prevailing adsorption mechanism is based on the structural similarities of CMC and cellulose.57 The adsorbed amount and thickness of the adsorbed layer were 141 ng cm−2 and 0.9 nm, respectively. These values are in agreement with previous reports.35,58 Next, the CMC-modified cellulose was activated by EDC–NHS solution to convert the carboxyl groups to amine-reactive esters. This activation step can be observed as a small increase in the SPR signal. After the EDC–NHS activation, anti-HSA affibody molecules were conjugated (via amide bonds) onto the activated CMC–cellulose surface. The conjugated amount and thickness of the anti-HSA layer were measured to be 84.7 ng cm−2 and 0.62 nm, respectively. The conjugated amount of anti-HSA affibodies was significantly lower than that of monoclonal antibodies on carboxymethylated dextran (CMD) surfaces (800–1000 ng cm−2).59,60 However, it should be noted here that the molecular weight of an antibody is almost ten-fold larger when compared to that of an affibody molecule (150 vs. 14 kDa). Moreover, it is likely that the differences in the surface construction, i.e., three-dimensional, highly hydrated CMC-modified cellulose surfaces compared to that of cellulose, affect protein adsorption. As a reference, the amount of anti-HSA bound onto the fully covered flat cellulose surface was calculated to be approximately 80 ng cm−2 if a closed packed monolayer of spherical anti-HSA (3 nm dimer radius and Mw of 14 kDa)61 is assumed. Therefore, it can be concluded that the achieved experimental conjugation level (84.7 ng cm−2) of anti-HSA was very satisfactory. It should be mentioned, that no adsorption was observed when the conjugation of anti-HSA was conducted in the absence EDC–NHS (Fig. 6b). Ethanol amine treatment was used as a last step, before challenging the prepared biointerface with HSA, in order to remove any unreacted NHS–esters. | ||
| Fig. 6 SPR sensograms corresponding to the conjugation of anti-HSA affibodies onto CMC-modified cellulose with (a) and without (b) EDC–NHS-mediated coupling. Subsequently, the binding of HSA on the respective surface are also shown in (c) and (d). | ||
Specific binding of HSA on the prepared anti-HSA affibody biointerfaces was approximately eight-fold higher (∼81 vs. 10 ng cm−2) when anti-HSA was conjugated onto cellulose via EDC–NHS chemistry (Fig. 6c and d). This fact highlights the importance of EDC–NHS coupling for improving the efficiency of the biointerface. Moreover, the detected amount of HSA (81 ng cm−2) is in good agreement with the amount of anti-HSA conjugated on cellulose, which indicates the high specificity of the system. CMC modification not only allows chemical conjugation of anti-HSA affibodies but reduces non-specific adsorption. For example, HSA adsorption on CMC-modified cellulose was 10 ng cm−2, more than five-fold lower than that onto pure cellulose, 57 ng cm−2 (Fig. S1, ESI†).
Filtration of HSA with anti-HSA affibody functionalized BC tubes
As a demonstration of the effectiveness of BC carrying affibodies for biofiltration, anti-HSA were immobilized onto the synthesized CMC–BC and TEMPO-oxidized BC tubes via EDC–NHS chemistry. It is important to note here that unwanted diffusion of anti-HSA through the wall of a BC tube may occur because of the small size of the affibody (14 kDa). Therefore, some of the conjugated affibodies are expected not to be available for binding with the target protein (HSA). In contrast, diffusion is not expected for the relatively large HSA molecules (66 kDa).20 Detection studies were carried out by using dansyl-stained HSA via fluorescence microscopy. The fluorescence imaging was conducted on the inner plane view of the BC-tubes noting that the fluorescence of a non-conjugated, anti-HSA-free CMC–BC tube was null (Fig. 7a). When non-conjugated, anti-HSA-free CMC–BC tubes were exposed to dansylated HSA a slight fluorescence was observed (Fig. 7b). This is explained by the small non-specific adsorption of HSA (see Fig. 6, SPR data). As expected, the fluorescence was significantly increased when HSA was filtrated through the CMC–BC tube containing the conjugated anti-HSA affibodies (Fig. 7c). This demonstrates that the conjugation of affibodies enhances the affinity and specificity of CMC–BC tubes to capture the target molecules. The normalized fluorescence values of CMC–BC tubes functionalized with anti-HSA affibody as a function of the HSA concentration are presented in Fig. 7d. The fluorescence intensity of bound HSA was found to increase linearly with HSA concentration in solution. The detection limit of HSA with CMC–BC tubes functionalized with anti-HSA was lower than 0.001 g L−1, which supports CMC–BC tubes as platform for specific biofiltration assays. However, it should be noted that further investigations are required to reveal the separation capacity limits of CMC–BC assays. The HSA filtration tests were repeated also with TEMPO-oxidized BC-tubes functionalized with anti-HSA affibody via EDC–NHS chemistry. The fluorescence intensities of bound dansylated HSA was found to be lower compared to that of anti-HSA affibody functionalized CMC–BC tubes (Fig. 7d). Moreover, the slope of the fluorescence intensity of bound HSA was significantly lower compared to that on CMC–BC, indicating a more limited biofiltration efficiency. The higher detection efficiency of anti-HSA affibody functionalized CMC–BC-tubes is most probably due to the higher carboxyl content of CMC–BC (Fig. 4a and b) and the hydrogel like adsorption layer of CMC on BC-fibrils, which may lead to higher surface concentration (more conjugation sites on BC) and higher accessibility of immobilized anti-HSA affibody on BC-fibrils. The SEM image of CMC–BC tubes functionalized with anti-HSA (Fig. 7e) was similar to that of pure CMC–BC tube (Fig. 2b and 3b and d). This can be taken as an indication of an even distribution of anti-HSA affibodies on the surface of CMB–BC tubes. The selective binding of HSA in the presence of proteins other than HSA was not attempted here but is the subject of on-going investigations. Altogether, the covalent conjugation of affibodies onto the surface of BC tubes and subsequent detection of a target protein was demonstrated. It is expected that the developed mild and generic methodology can be effortlessly transformed and utilized for the specific detection and separation of other target proteins. | ||
| Fig. 7 Detection of HSA in BC tubes functionalized with anti-HSA by using fluorescence imaging. Included are the surface of a CMC–BC tube free of anti-HSA before (a) and after (b) exposure to dansylated HSA. The image in (c) corresponds to the surface after exposure to dansylated HSA to CMC–BC tube conjugated with anti-HSA affibodies. The normalized fluorescence of dansylated HSA as a function of HSA concentration on anti-HSA biointerfaces prepared on the CMC-modified and TEMPO-oxidized BC tubes is included in (d). A SEM plane view image of the inner surface of a CMC–BC tube functionalized with anti-HSA is also shown (e). | ||
Conclusions
The synthesis of BC–CMC modified tubes and their functionalization by affibody conjugation is demonstrated. The presence of CMC in the culture medium during BC synthesis has a significant influence in the WRV of never-dried and air-dried BC tubes and reduces irreversible structural changes during drying of BC. In addition, BC activation with CMC improves the removal of protein residues from the synthesized cellulose and facilitates anti-human serum albumin (anti-HSA) conjugation by covalently binding the affibody to the carboxyl groups via EDC–NHS coupling. CMC modification after alkaline TEMPO-oxidation of BC resulted in a carboxyl group density lower than that of CMC–BC. TEMPO-oxidized BC did not prevent irreversible structural changes to the same extent than the CMC-modification. The specific binding of HSA onto anti-HSA ligands supported on the BC interface was demonstrated by SPR. Finally, CMC-modified and TEMPO-oxidized BC tubes functionalized with anti-HSA were used to capture fluorescent HSA from solution. The fluorescence intensity of bound HSA on both types of BC tubes increased linearly as a function of HSA concentration. However, CMC–BC tubes carrying conjugated anti-HSA affibodies were more effective in binding HSA compared to TEMPO-oxidized BC tubes with conjugated anti-HSA. It is expected that the presented generic and robust method for grafting recombinant affibody proteins onto BC materials has a potential to open up new venues in the field of biofiltration, i.e., selective separation and detection of various target molecules from an analyte solution.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Funding sources
This work has been performed as a part of the Lignocell project of the Finnish Centre for Nanocellulosic Technologies financially supported by the Finnish Funding Agency for Technology and Innovation (TEKES) and UPM. Furthermore, this work was carried out under the Academy of Finland’s Centres of Excellence Programme (2014-2019).Abbreviations
| BC | Bacterial cellulose |
| CMC | Carboxymethyl cellulose |
| HSA | Human serum albumin |
| SPR | Surface plasmon resonance |
| XPS | X-rays photoelectron spectroscopy |
Acknowledgements
We thank Dr Joseph M. Campbell for performing XPS measurements, and Anu Anttila, Ritva Kivelä, and Marja Kärkkäinen for technical assistance.References
- D. Klemm, F. Kramer, S. Moritz, T. Lindström, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef CAS PubMed.
- Y. Zhang, T. Nypelo, C. Salas, J. Arboleda, I. C. Hoeger and O. J. Rojas, J. Renewable Mater., 2013, 1, 195–211 CrossRef CAS.
- M. Pääkkö, M. Ankerfors, H. Kosonen, A. Nykänen, S. Ahola, M. Österberg, J. Ruokolainen, J. Laine, P. T. Larsson, O. Ikkala and T. Lindström, Biomacromolecules, 2007, 8, 1934–1941 CrossRef PubMed.
- K. Syverud and P. Stenius, Cellulose, 2009, 16, 75–85 CrossRef CAS.
- H. Orelma, I. Filpponen, L. Johansson, M. Österberg, O. Rojas and J. Laine, Biointerphases, 2012, 7, 1–12 CrossRef PubMed.
- Y. Zhang, R. G. Carbonell and O. J. Rojas, Biomacromolecules, 2013, 14, 4161–4168 CrossRef CAS PubMed.
- R. Jonas and L. F. Farah, Polym. Degrad. Stab., 1998, 59, 101–106 CrossRef CAS.
- R. E. Cannon and S. M. Anderson, Crit. Rev. Microbiol., 1991, 17, 435–447 CrossRef CAS PubMed.
- M. Iguchi, S. Yamanaka and A. Budhiono, J. Mater. Sci., 2000, 35, 261–270 CrossRef CAS.
- W. Borzani and S. Souza, Biotechnol. Lett., 1995, 17, 1271–1272 CrossRef CAS.
- W. Tang, S. Jia, Y. Jia and H. Yang, World J. Microbiol. Biotechnol., 2010, 26, 125–131 CrossRef CAS.
- S. Yamanaka, K. Watanabe, N. Kitamura, M. Iguchi, S. Mitsuhashi, Y. Nishi and M. Uryu, J. Mater. Sci., 1989, 24, 3141–3145 CrossRef CAS.
- W. K. Czaja, D. J. Young, M. Kawecki and R. M. Brown, Biomacromolecules, 2007, 8, 1–12 CrossRef CAS PubMed.
- M. Onodera, I. Harashima, K. Toda and T. Asakura, Biotechnol. Bioprocess Eng., 2002, 12, 289–294 CrossRef.
- A. Bodin, H. Bäckdahl, H. Fink, L. Gustafsson, B. Risberg and P. Gatenholm, Biotechnol. Bioeng., 2007, 97, 425–434 CrossRef CAS PubMed.
- G. Helenius, H. Bäckdahl, A. Bodin, U. Nannmark, P. Gatenholm and B. Risberg, J. Biomed. Mater. Res., Part A, 2006, 76, 431–438 CrossRef PubMed.
- D. Klemm, D. Schumann, U. Udhardt and S. Marsch, Prog. Polym. Sci., 2001, 26, 1561–1603 CrossRef CAS.
- W. Czaja, A. Krystynowicz, S. Bielecki and R. M. Brown Jr, Biomaterials, 2006, 27, 145–151 CrossRef CAS PubMed.
- A. M. Sokolnicki, R. J. Fisher, T. P. Harrah and D. L. Kaplan, J. Membr. Sci., 2006, 272, 15–27 CrossRef CAS PubMed.
- H. Shibazaki, S. Kuga, F. Onabe and M. Usuda, J. Appl. Polym. Sci., 1993, 50, 965–969 CrossRef CAS.
- F. Rusmini, Z. Zhong and J. Feijen, Biomacromolecules, 2007, 8, 1775–1789 CrossRef CAS PubMed.
- R. Pelton, TrAC, Trends Anal. Chem., 2009, 28, 925–942 CrossRef CAS PubMed.
- A. Isogai and Y. Kato, Cellulose, 1998, 5, 153–164 CrossRef CAS.
- W. Shen, S. Chen, S. Shi, X. Li, X. Zhang, W. Hu and H. Wang, Carbohydr. Polym., 2009, 75, 110–114 CrossRef CAS PubMed.
- D. Klemm, B. Philipp and T. Heinze, Comprehensive cellulose chemistry. Vol. 2, Functionalization of cellulose, VCH, Weinheim, 1998, p. 389 Search PubMed.
- S. E. C. Whitney, J. E. Brigham, A. H. Darke, J. S. G. Reid and M. J. Gidley, Plant J., 1995, 8, 491–504 CAS.
- C. H. Haigler, A. R. White, R. M. Brown and K. M. Cooper, J. Cell Biol., 1982, 94, 64–69 CrossRef CAS.
- H. T. Winter, C. Cerclier, N. Delorme, H. Bizot, B. Quemener and B. Cathala, Biomacromolecules, 2010, 11, 3144–3151 CrossRef CAS PubMed.
- D. Ciechanska, Fibres Text. East. Eur., 2004, 12, 69–72 CAS.
- Q. Zhou, E. Malm, H. Nilsson, P. T. Larsson, T. Iversen, L. A. Berglund and V. Bulone, Soft Matter, 2009, 5, 4124–4130 RSC.
- E. E. Brown and M. G. Laborie, Biomacromolecules, 2007, 8, 3074–3081 CrossRef CAS PubMed.
- M. Seifert, S. Hesse, V. Kabrelian and D. Klemm, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 463–470 CrossRef CAS.
- P. Nygren, FEBS J., 2008, 275, 2668–2676 CrossRef CAS PubMed.
- H. A. Shelanski and A. M. Clark, J. Food Sci., 1948, 13, 29–35 CrossRef CAS PubMed.
- H. Orelma, T. Teerinen, L. Johansson, S. Holappa and J. Laine, Biomacromolecules, 2012, 13, 1051–1058 CrossRef CAS PubMed.
- C. Castro, R. Zuluaga, C. Álvarez, J. Putaux, G. Caro, O. J. Rojas, I. Mondragon and P. Gañán, Carbohydr. Polym., 2012, 89, 1033–1037 CrossRef CAS PubMed.
- S. Hestrin and M. Schramm, Biochem. J., 1954, 58, 345–352 CAS.
- L. Johansson and J. M. Campbell, Surf. Interface Anal., 2004, 36, 1018–1022 CrossRef CAS.
- G. Beamson and D. Briggs, High resolution XPS of organic polymers, Wiley, Chichester, UK, 1992, p. 295 Search PubMed.
- J. V. Staros, R. W. Wright and D. M. Swingle, Anal. Biochem., 1986, 156, 220–222 CrossRef CAS.
- B. Ronald, J. Pharmacol. Toxicol. Methods, 2001, 45, 247–253 CrossRef.
- L. S. Jung, C. T. Campbell, T. M. Chinowsky, M. N. Mar and S. S. Yee, Langmuir, 1998, 14, 5636–5648 CrossRef CAS.
- S. Trabelsi and D. Langevin, Langmuir, 2006, 23, 1248–1252 CrossRef PubMed.
- F. W. Putnam, in The Plasma Proteins: Structure, Function and Genetic Control, Academic press, New York, 1984, vol. 2, p. 420 Search PubMed.
- M. Rinaudo and M. Mils, Biopolymers, 1978, 17, 2663–2678 CrossRef CAS.
- T. Tammelin, T. Saarinen, M. Oesterberg and J. Laine, Cellulose, 2006, 13, 519–535 CrossRef CAS PubMed.
- C. Aulin, S. Ahola, P. Josefsson, T. Nishino, Y. Hirose, M. Österberg and L. Wagberg, Langmuir, 2009, 25, 7675–7685 CrossRef CAS PubMed.
- M. Schaub, G. Wenz, G. Wegner, A. Stein and D. Klemm, Adv. Mater., 1993, 5, 919–922 CrossRef CAS.
- G. Ben-Hayyim and I. Ohad, J. Cell Biol., 1965, 25, 191–207 CrossRef CAS.
- C. J. Grande, F. G. Torres, C. M. Gomez and M. Carmen Bañó, Acta Biomater., 2009, 5, 1605–1615 CrossRef CAS PubMed.
- T. Saito and A. Isogai, Biomacromolecules, 2004, 5, 1983–1989 CrossRef CAS PubMed.
- Y. Okita, T. Saito and A. Isogai, Biomacromolecules, 2010, 11, 1696–1700 CrossRef CAS PubMed.
- S. Ifuku, M. Tsuji, M. Morimoto, H. Saimoto and H. Yano, Biomacromolecules, 2009, 10, 2714–2717 CrossRef CAS PubMed.
- T. Saito, M. Yanagisawa and A. Isogai, Cellulose, 2005, 12, 305–315 CrossRef CAS PubMed.
- J. DiFlavio, R. Pelton, M. Leduc, S. Champ, M. Essig and T. Frechen, Cellulose, 2007, 14, 257–268 CrossRef CAS.
- L. Johansson, T. Tammelin, J. M. Campbell, H. Setala and M. Osterberg, Soft Matter, 2011, 7, 10917–10924 RSC.
- J. Laine, T. Lindstrom, G. G. Nordmark and G. Risinger, Nord. Pulp Pap. Res. J., 2000, 15, 520–526 CAS.
- H. Orelma, I. Filpponen, L. Johansson, J. Laine and O. J. Rojas, Biomacromolecules, 2011, 12, 4311–4318 CrossRef CAS PubMed.
- B. Johnsson, S. Löfås and G. Lindquist, Anal. Biochem., 1991, 198, 268–277 CrossRef CAS.
- F. Yu, B. Persson, S. Löfas and W. Knoll, Anal. Chem., 2004, 76, 6765–6770 CrossRef CAS PubMed.
- C. Eigenbrot, M. Ultsch, A. Dubnovitsky, L. Abrahmsén and T. Härd, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15039–15044 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SPR curves for the adsorption of HSA on pure, CMC-modified, and anti-HSA functionalized CMC-modified cellulose. See DOI: 10.1039/c4ra08882d |
| This journal is © The Royal Society of Chemistry 2014 |