
Four cobalt(ii) coordination polymers with diverse topologies derived from flexible bis(benzimidazole) and aromatic dicarboxylic acids: syntheses, crystal structures and catalytic properties†
Abstract
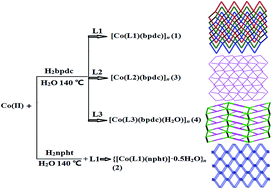
Four mixed ligand coordination polymers based on the flexible bis(5,6-dimethybenzimidazole) and aromatic dicarboxylic acids, namely, [Co(L1)(bpdc)]n (1), {[Co(L1)(npht)]·0.5H2O}n (2), [Co(L2)(bpdc)]n (3), and [Co(L3)(bpdc)(H2O)]n (4) (L1 = 1,4-bis(5,6-dimethylbenzimidazol-1-ylmethyl)benzene, H2bpdc = 4,4′-biphenyldicarboxylic acid, L2 = 1,3-bis(5,6-dimethylbenzimidazol-1-ylmethyl)benzene, H2npht = 3-nitrophthalic acid, L3 = 1,1′-bis(5,6-dimethylbenzimidazole)methane) have been hydrothermally synthesized and structurally characterized. Polymer 1 features a 3D three-fold interpenetrating dia array with a 4-connected 66 network, while 2 exhibits a 3D noninterpenetrated 3-connected framework with a 103-ThSi2 architecture. 3 and 4 have two-dimensional 3-connected (63) and 4-connected (44.62) topologies, respectively. Complex 4 ultimately is extended into an unusual 3D (3,5)-connected seh-3,5-P21/c supramolecular network via O–H⋯O hydrogen bonding interactions. The fluorescence and catalytic properties of the complexes for the degradation of the Congo red azo dye in a Fenton-like process are reported.
 Please wait while we load your content...
Please wait while we load your content...

