
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolid Lewis acidity of boehmite γ-AlO(OH) and its catalytic activity for transformation of sugars in water
Atsushi
Takagaki
*a,
Ji Chul
Jung
b and
Shigenobu
Hayashi
c
aDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: atakagak@chemsys.t.u-tokyo.ac.jp
bDepartment of Chemical Engineering, Myongji University, Yongin 449-728, South Korea
cResearch Institute of Instrumentation Frontier, National Institute of Advanced Industrial Science and Technology (AIST), Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan
First published on 5th September 2014
Abstract
Aqueous-phase transformation of C3- and C6-sugars toward valuable intermediates such as lactic acid and 5-hydroxymethylfurfural (HMF) using boehmite, an abundant, inexpensive and simple aluminium oxide hydroxide γ-AlO(OH) was demonstrated. Pyridine-adsorbed Fourier transform infrared (FTIR) spectroscopy and trimethylphosphine oxide-adsorbed 31P magic angle spinning (MAS) nuclear magic resonance (NMR) revealed that boehmite had no Brønsted acid sites and a small amount of weak Lewis acid sites. The amount of Lewis acid sites and the initial reaction rate for lactic acid synthesis from dihydroxyacetone increased with increase of calcination temperature of boehmite whereas the selectivity for lactic acid remained unchanged and turnover frequency decreased. Boehmite afforded lactic acid selectively from aqueous C3-sugars solution with the yields of 32% and 42% from dihydroxyacetone and glyceraldehyde, respectively, which were higher than those of hydrotalcite, titanium oxide, tin oxide and tungsten oxide. Moreover, boehmite gave both lactic acid and HMF simultaneously from aqueous glucose solution as well as fructose. The total yields of lactic acid and HMF from aqueous glucose solution reached 40%. Boehmite was found to catalyse a variety of reactions including isomerization, retro-aldol condensation, dehydration and hydration in water.
Introduction
Catalytic conversion of biomass derivatives has received much attention. One of the most fascinating processes is aqueous-phase transformation of carbohydrates towards important intermediates such as lactic acid and 5-hydroxymethylfurfural (HMF) using heterogeneous catalysts. Lewis acid is highly effective for selective production of lactic acid from triose and hexose in water,1 and some solid Lewis acid catalysts such as metallosilicate zeolites (Sn-, Zr-, Ti-beta),2,3 H-USY zeolite (Si/Al = 6)4 and desilicated ZSM-5 zeolite5 have been demonstrated. Tin-beta catalyst afforded 90% yield of lactic acid from triose in water at 353 K for 24 h whereas Zr-beta and Ti-beta gave 44 and 25% yield of lactic acid, respectively.2 A tin-containing porous carbon–silica composite material (Sn–Si–CSM) was also effective for lactic acid synthesis. The catalyst gave 92% conversion of dihydroxyacetone (DHA) and 83% selectivity of lactic acid in water at 383 K for 6 h.6 A high aluminum H-USY zeolite (Si/Al = 6) afforded 100% conversion of DHA and 71% selectivity of lactic acid at 398 K for 24 h.4 Recently, it was reported that a hierarchical ZSM5 catalyst synthesized by desilication of commercial MFI-type zeolite gave 90% conversion of DHA and 91% selectivity of lactic acid in water at 413 K for 6 h.5 Lewis acid also gives HMF from hexose in water or biphasic system, and different types of solid Lewis acid catalysts such as amorphous niobium oxide hydrate (Nb2O5·nH2O),7 tantalum oxide hydrate (Ta2O5·nH2O),8 titanium oxide,9 and mesoporous titanium oxide10 have been reported.Aluminium is one of the most abundant elements with 3rd Clark number and possesses Lewis acidity in nature represented as AlCl3. Aluminium oxide is widely used as a solid Lewis acid catalyst for a variety of reactions including dehydration, isomerization and alkylation. Alumina (γ-Al2O3) has been also employed as a support of precious metal for aqueous-phase transformation of cellulose in the early study of biomass conversion over heterogeneous catalyst.11,12 However, it is well known that γ-Al2O3 is unstable in hot water and is easily converted into an aluminium oxide hydroxide, boehmite (γ-AlO(OH)) above 423 K.13 In many cases, aqueous-phase transformation of sugars using heterogeneous catalyst has been carried out above 423 K. Thus, the use of boehmite as a solid acid catalyst would be desirable.
Here, we examined solid acid properties of boehmite and its catalytic activity for transformation of triose and hexose in hot water. To the best of our knowledge, this is the first successful example of using boehmite as a solid Lewis acid catalyst for transformation of sugars in aqueous solution.
Boehmite has a two-dimensional structure composed of AlO6 octahedra as shown in Fig. 1. All OH groups are exposed on the surface of the octahedral double layers.
 | ||
| Fig. 1 Crystal structure of boehmite. | ||
A representative reaction pathway of transformation of sugars is shown in Scheme 1. Glucose is converted into fructose via isomerization catalysed by Lewis acid. HMF can be obtained from fructose via dehydration by removal of three water molecules. From these hexoses, trioses such as glyceraldehyde and dihydroxyacetone are formed via retro-aldol condensation which is also catalysed by Lewis acid. These trioses can be interconverted via isomerization. Pyruvaldehyde is formed by dehydration of these trioses, which is further transformed into lactic acid via a 1,2-hydride shift, which is also catalysed by Lewis acid. Therefore Lewis acid sites play an important role for transformation of sugars.
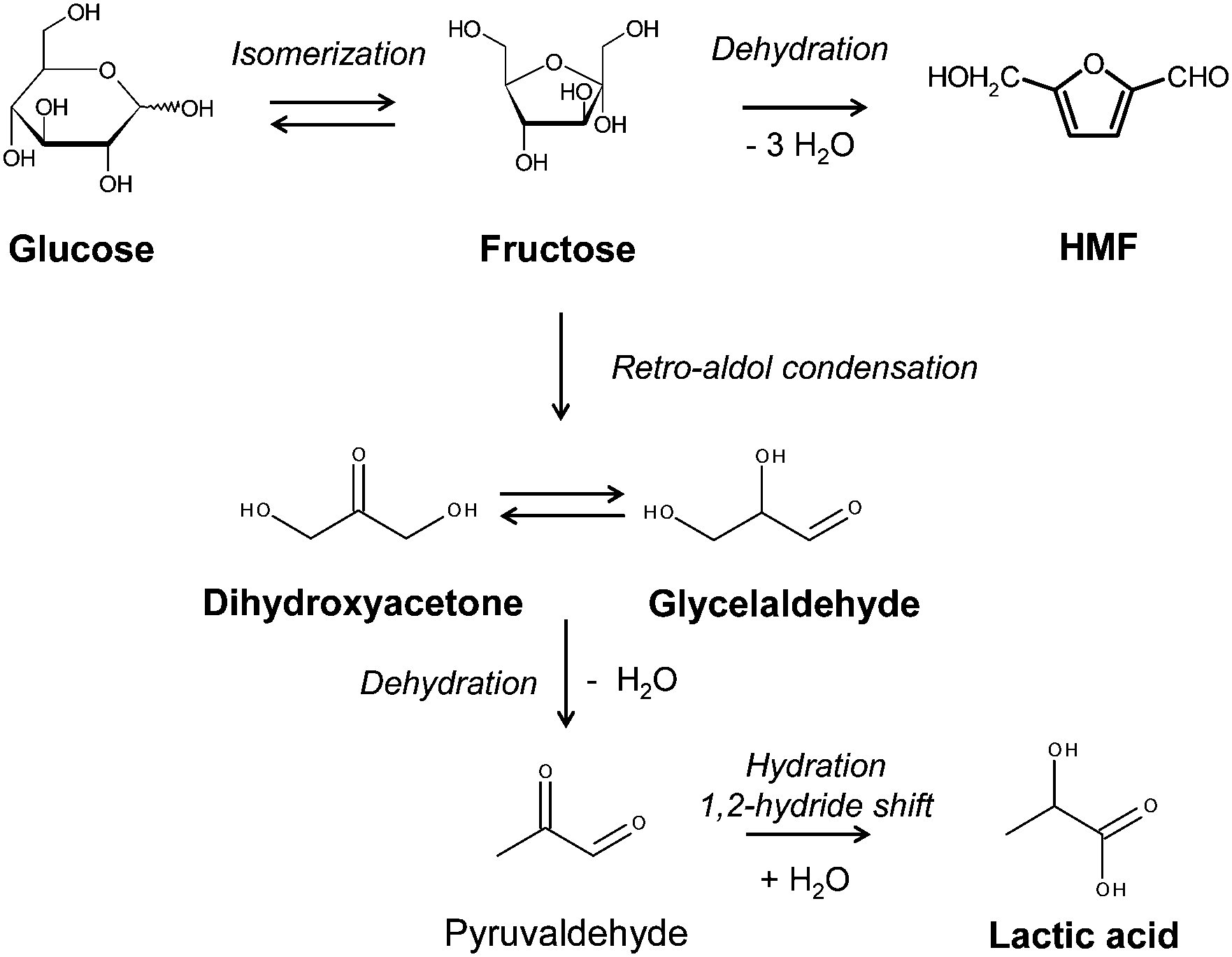 | ||
| Scheme 1 A representative reaction pathway for transformation of hexose and triose. | ||
Experimental
Materials
Boehmite, TiO2, SnO2 and WO3 were purchased from Wako Pure Chemical Industries. Alumina was obtained from Sigma-Aldrich (γ-Al2O3, #544833). Boehmite was calcined at 453, 573 and 773 K for 5 h in air. Niobic acid (Nb2O5·nH2O) was supplied from Companhia Brasileira de Metalurgia e Mineração (CBMM). Mg–Al hydrotalcite (Mg/Al = 3) was prepared by a conventional coprecipitation method according to the literature.14 Dihydroxyacetone dimer (98%), DL-glyceraldehyde (97%), D(+)-glucose (98%) and D(−)-fructose (99%) were purchased from Wako Pure Chemical Industries.Characterization
The samples were characterized by X-ray diffraction (XRD; RINT-2700, Rigaku), scanning electron microscopy (SEM; S-4700, Hitachi) and nitrogen adsorption–desorption (BELSORP-miniII, BEL Japan). XRD patterns of the samples were obtained with the diffractometer operated at 40 kV and 100 mA, using Cu-Kα radiation. Nitrogen adsorption–desorption isotherms were measured at 77 K. The samples were pretreated at 453 K for 1 h under vacuum. The Brunauer–Emmett–Teller (BET) surface area was estimated over a relative pressure range of 0.05–0.30. The pore size distribution was calculated from analysis of the adsorption branches of the isotherms using the Barrett–Joyner–Halenda (BJH) method.Acid property of the samples was examined using pyridine-adsorbed Fourier transform infrared (FTIR) spectroscopy and trimethylphosphine oxide (TMPO)-adsorbed 31P magic angle spinning (MAS) nuclear magic resonance (NMR). In FTIR measurement, the samples were prepared in the form of self-supporting disc and placed in a conventional gas-circulation system. The samples were pretreated at 453 K for 1 h under vacuum. Then, pyridine was introduced into the system at 373 K. After 30 min, the samples were evacuated for 30 min to remove gas phase pyridine. The IR spectra before and after adsorption of pyridine were recorded by using the spectrometer (FT/IR-6100, Jasco) equipped with a MCT detector with a resolution of 4 cm−1. The amount of Lewis acid sites was determined on the basis of the integral absorbance of the characteristic band at 1450 cm−1 by using integrated molar extinction coefficients, 2.22 cm μmol−1.15
In NMR measurement, the TMPO-adsorbed samples were prepared by evacuation at 453 K for 1 h followed by immersion in dichloromethane solution containing 0.2 M TMPO at room temperature for 1 day in a glovebox under nitrogen. 0.06 mmol of TMPO was exposed to 0.3 g of boehmite. After evacuation to remove the CH2Cl2 solvent, the samples were packed in a rotor housed in a glovebox with an N2 atmosphere. 31P MAS NMR spectra were measured at room temperature using Bruker Avance 400 spectrometer at Larmor frequency of 162.0 MHz. Bruker MAS probehead was used in combination with a 4 mm zirconia rotor operated at a sample spin rate of 10 kHz. The single pulse sequence in combination with 1H high power decoupling during signal acquisition was used with a π/4 pulse and a repetition time of 3 s. The 31P chemical shift was referenced to 85% H3PO4 at 0.0 ppm. (NH4)2HPO4 was used as a second reference material with signal set at 1.33 ppm.
Transformation of sugars
A quantity of 0.28 mmol substrate with 50 mg of solid catalyst was stirred in 3 mL water in a glass reactor vessel (Ace glass pressure tube). Dihydroxyacetone dimer, glyceraldehyde, glucose and fructose were used as reactants. The reactor vessel was placed in an oil bath at 353–443 K. The reactants and products were analysed by high performance liquid chromatography (HPLC; LC-2000 plus, Jasco) equipped with a differential refractive index detector (RI-2031 plus, Jasco) using Aminex HPX-87H column (flow rate: 0.5 mL min−1, eluent: H2SO4 10 mM).Results and discussion
Structure of boehmite calcined at different temperatures
X-ray diffraction patterns for boehmite samples calcined at different temperatures for 5 h are shown in Fig. 2. The samples calcined at 453 and 573 K showed four distinct peaks located at 2θ = 14.3°, 28.2°, 38.2° and 49.0°, which are assigned to the (020), (120), (031) and (051) phases of boehmite, respectively. The peak intensities and widths remained unchanged up to 573 K calcination. The crystallite sizes were estimated to be 4.5 nm for both samples according to Scherrer equation D = Kλ/(β cos θ) where K, λ, β and θ are the shape factor (0.9), X-ray wavelength (0.154 nm), the line broadening at half the maximum intensity in radians, and the Bragg angle, respectively. Further calcination caused phase change from boehmite to γ-alumina. The sample calcined at 773 K showed different diffraction patterns from other two samples. The peaks at 45.9° and 66.6° correspond to the (400) and (440) phases of γ-alumina. No peaks for boehmite were observed. This result is in good agreement with previous studies on the thermal transformation of boehmite in which the phase change to γ-alumina takes place above ca. 723 K.13,16 | ||
| Fig. 2 X-ray diffraction patterns for boehmite samples calcined at 453, 573 and 773 K. | ||
The changes of morphology were observed by using scanning electron microscopy (SEM) (Fig. 3). A plate-like structure in size below 100 nm was observed for the samples calcined at 453 and 573 K, which is characteristic to layered boehmite. In contrast, such two-dimensional structure was not seen for the sample calcined at 773 K because of phase transformation from boehmite to γ-alumina. For all samples, small particles were aggregated to form secondary particles.
 | ||
| Fig. 3 SEM images of boehmite samples calcined at 453 K, 573 K and 773 K. | ||
The nitrogen sorption isotherms and pore size distributions are shown in Fig. 4. The BET surface areas and pore volumes are listed in Table 1. The amount of N2 sorption at low relative pressure region depended little on calcination temperature. The BET surface areas were 257, 213 and 229 m2 g−1 for the samples calcined at 453, 573 and 773 K, respectively. In contrast, the amount of N2 sorption at high relative pressure region increased with increase of calcination temperatures, indicating the presence of mesopores due to the formation of void spaces between particles. The pore volume increased from 0.45 to 0.61 mL g−1. The samples calcined at 453 and 573 K did not have a characteristic peak in the differential pore size distributions whereas the sample calcined at 773 K had a peak at 2.7 nm.
 | ||
| Fig. 4 (a) N2 sorption isotherms and (b) pore size distributions of boehmite samples calcined at 453, 573 and 773 K. | ||
Characterization of acid sites
The solid acid property was investigated by using pyridine-adsorbed Fourier transform infrared spectroscopy (Py-FTIR) and trimethylphosphine oxide-adsorbed 31P magic angle spinning nuclear magnetic resonance (31P MAS NMR). The Py-FTIR spectroscopy is a useful and conventional method to identify Brønsted and Lewis character of solid acids. The FTIR spectra of calcined boehmite samples are shown in Fig. 5. The bands at 1615, 1577, 1492 and 1450 cm−1 appeared for three samples, which are assigned to coordinately bonded pyridine adsorbed on Lewis acid sites of samples. No band at 1540 cm−1 was observed, indicating the absence of Brønsted acid sites. The intensities of these bands increased with increase of calcination temperature, but no other significant changes such as band shifts and generation of Brønsted acid sites were observed in spite of the crystal phase transformation. The amount of Lewis acid sites was estimated on the basis of the absorbance of band at 1450 cm−1 (Table 1). The amount of Lewis acid sites of the sample calcined at 453 K was 50 μmol g−1. This is close value to that of the literature in which boehmite was obtained from γ-alumina by hydrothermal treatment (42 μmol g−1).13 The amount of Lewis acid sites of the samples calcined at 573 and 773 K were 109 and 218 μmol g−1, respectively. The acid site densities (sites per nm2) calculated by using the amount of Lewis acid sites and the BET surface areas are also listed in Table 1. These were 0.12, 0.31 and 0.57 acid sites per nm2 for samples calcined at 453, 573 and 773 K, respectively. According to the crystal structure of boehmite (a = 0.369 nm, c = 0.286 nm), it is considered that 9.48 Al per nm2 are exposed on the surface. Assuming that the Lewis acid sites were formed on the surface of boehmite, only 1–3% of the surface Al exhibited Lewis acidity. Alternatively, if the percentage of Al that shows Lewis acidity was calculated from whole Al contents based on molecular formula, then it was found that 0.3, 0.7 and 1.1 atom% of Al functioned as Lewis acid sites for samples calcined at 453, 573 and 773 K, respectively. | ||
| Fig. 5 FTIR spectra of pyridine adsorbed on boehmite samples calcined at 453, 573 and 773 K. | ||
Next, the acid strength of Lewis acid sites of samples was examined by solid-state NMR. Although the method of temperature-programmed desorption of ammonia (NH3-TPD) is a conventional technique to determine both acid strength and amount of solid catalysts, this is not applicable for the samples whose crystal structures and acid properties could be changed during raising temperature. In contrast, the solid-state NMR using trimethylphosphine oxide (TMPO) as a probe molecule can be measured at room temperature and provides useful information on acid strength in terms of chemical shift, δ. The resonance peaks with high chemical shifts reflect strong acidity. For instance, the peaks for very strong Lewis acid sites of sulphated zirconia were found at 90 and 63 ppm.17 Those for strong Lewis acid sites of scandium triflate (Sc(OTf)3)18 and weak Lewis acid sites of silica–alumina19 were observed at 64.5 and 53 ppm, respectively. The 31P MAS NMR spectra of calcined boehmite samples are shown in Fig. 6. The sample calcined at 453 K exhibited a peak at 54 ppm accompanying the main peak at 44 ppm, attributable to weak Lewis acid sites. Thus, the Lewis acid strength of boehmite was weak and comparable to that of silica–alumina. The large peak at 44 ppm can be assigned to physisorbed TMPO19 or very weak acid sites of Al–OH because the peaks at 40–41 ppm were assigned to silanols of zeolites.20,21 Thus, the large peak at 44 ppm is not related to Lewis acid sites. The peaks attributed to weak Lewis acid sites appeared at 48–49 ppm for the samples calcined at 573 and 773 K, indicating that the Lewis acid sites somewhat weakened by the heat treatment. Only the peaks at 48–54 ppm are attributed to Lewis acid sites. There was a good correlation between the peak areas around 48–54 ppm of 31P NMR and the peak areas at 1449 cm−1 of pyridine-FTIR. This clearly indicates that the peaks around 48–54 ppm of NMR are attributable to Lewis acid sites.
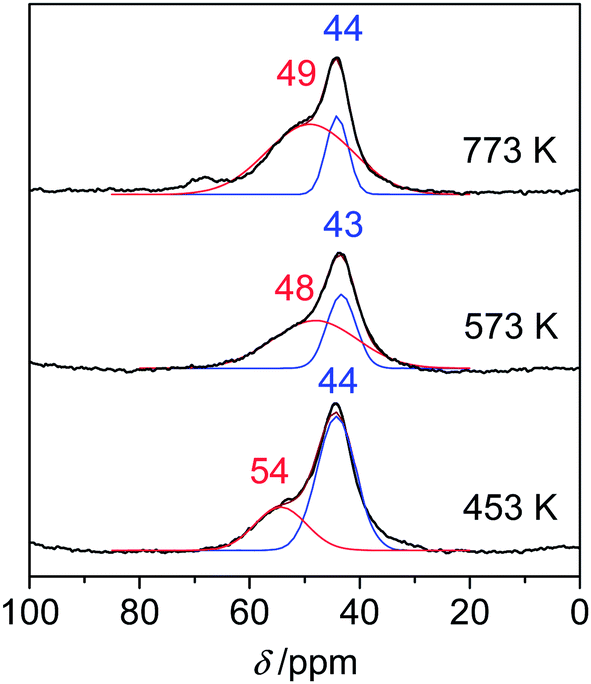 | ||
| Fig. 6 31P MAS NMR spectra for TMPO-adsorbed boehmite samples calcined at 453, 573 and 773 K. | ||
Acid catalytic activity for transformation of sugars in water
Transformation of triose was first investigated using the calcined boehmite samples. The results of transformation of dihydroxyacetone (DHA) in water over the three catalysts are listed in Table 2. Lactic acid was successfully formed over the three samples where the selectivity was almost the same (about 30%) at high conversions (87–96%). The initial reaction rate (r0) and turnover frequency (TOF) are also listed in Table 2. With increase of calcination temperature the initial reaction rate increased from 13.5 to 21.6 mmol g−1 h−1, turnover frequency, however, decreased from 270 to 99 h−1. The sample calcined at 453 K exhibited the highest TOF, which might be due to relatively stronger Lewis acidity. From these results, the sample calcined at 453 K was used for further investigation.| Calcination temp./K | Conv./% | Sel./% | r 0/mmol g−1 h−1e | TOF/h−1 | ||
|---|---|---|---|---|---|---|
| Glab | PAc | LAd | ||||
| a Reaction conditions: dihydroxyacetone dimer (0.28 mmol), catalyst (50 mg), water (3 mL), 413 K, 3 h. b Glyceraldehyde. c Pyruvaldehyde. d Lactic acid. e Initial reaction rate. | ||||||
| 453 | 87 | 1 | 3 | 28 | 13.5 | 270 |
| 573 | 90 | 0 | 2 | 31 | 15.0 | 138 |
| 773 | 96 | 0 | 2 | 29 | 21.6 | 99 |
The catalytic activity of boehmite was compared with a variety of metal oxides and hydroxides, and the results are shown in Table 3. Boehmite afforded lactic acid with 28% and 32% selectivity at 413 and 443 K, respectively (entries 1 and 2). The catalyst was recovered by centrifugation, washing with water, drying at 373 K in oven overnight, and recycled for further reaction. It was found that the boehmite was reusable and showed good lactic acid selectivity even after 3rd use (entries 3 and 4). Surprisingly, the selectivity of lactic acid was improved from 28% to 59%. After first reaction, the catalyst became brown due to the formation of humin. This brown colour remained unchanged after washing with water. It can be considered that the presence of humin on the catalyst suppressed the further formation of humin, resulting in the high selectivity of lactic acid. Alumina (γ-Al2O3) showed 91% conversion and 35% selectivity of lactic acid, which is slightly higher than boehmite (entry 6). Mg–Al hydrotalcite, a solid base catalyst, did not afford lactic acid (entry 7). Nb2O5·nH2O, a water-tolerant solid Lewis acid catalyst, gave high yield of lactic acid (entry 8). Other metal oxides such as TiO2, SnO2 and WO3 afforded pyruvaldehyde as an intermediate (entries 9–11), which is due to the lack of the activity of hydride transfer catalysed by Lewis acid sites. Thus, boehmite AlO(OH) composed of a common metal is a good candidate of Lewis acid catalyst for lactic acid production though Nb2O5·nH2O composed of a minor metal exhibited a superior activity.
| Entry | Catalyst | Conv./% | Sel./% | ||
|---|---|---|---|---|---|
| Glab | PAc | LAd | |||
| a Reaction conditions: dihydroxyacetone dimer (0.28 mmol), catalyst (50 mg), water (3 mL), 413 K, 3 h. b Glyceraldehyde. c Pyruvaldehyde. d Lactic acid. e Boehmite calcined at 453 K. f 433 K, 3 h. g 2nd use. h 3rd use. i Glyceraldehyde (0.28 mmol), catalyst (50 mg), water (3 mL), 433 K, 3 h. j Mg/Al = 3. | |||||
| 1 | AlO(OH)e | 87 | 1 | 3 | 28 |
| 2 | AlO(OH)f | >99 | 0 | 0 | 32 |
| 3 | AlO(OH)g | 98 | 4 | 10 | 42 |
| 4 | AlO(OH)h | 80 | 9 | 9 | 59 |
| 5 | AlO(OH)i | >99 | 0 | 0 | 42 |
| 6 | γ-Al2O3 | 91 | 1 | 1 | 35 |
| 7 | Mg–Al hydrotalcitej | >99 | 0 | 0 | 4 |
| 8 | Nb2O5·nH2O | >99 | 0 | 0 | 72 |
| 9 | TiO2 | 86 | 1 | 31 | 9 |
| 10 | SnO2 | 60 | 1 | 50 | 7 |
| 11 | WO3 | 81 | 0 | 17 | 0 |
The time-course of product distribution and dependence of product selectivity on DHA conversion are shown in Fig. 7. DHA was rapidly converted within 1.5 h (Fig. 7(a)). At the initial stage of the reaction, glyceraldehyde and pyruvaldehyde were formed. From the profiles of product selectivity (Fig. 7(b)), it was revealed that the formation of lactic acid from DHA is successive reactions, and primary, secondary and final products could be glyceraldehyde, pyruvaldehyde and lactic acid, respectively. However, the selectivity of glyceraldehyde at low DHA conversion was rather low. It can be considered that pyruvaldehyde was formed from not only glyceraldehyde but also DHA directly. When glyceraldehyde was used as reactant under the same reaction conditions as DHA, lactic acid was also formed with 42% selectivity at almost 100% conversion (Table 3, entry 5).
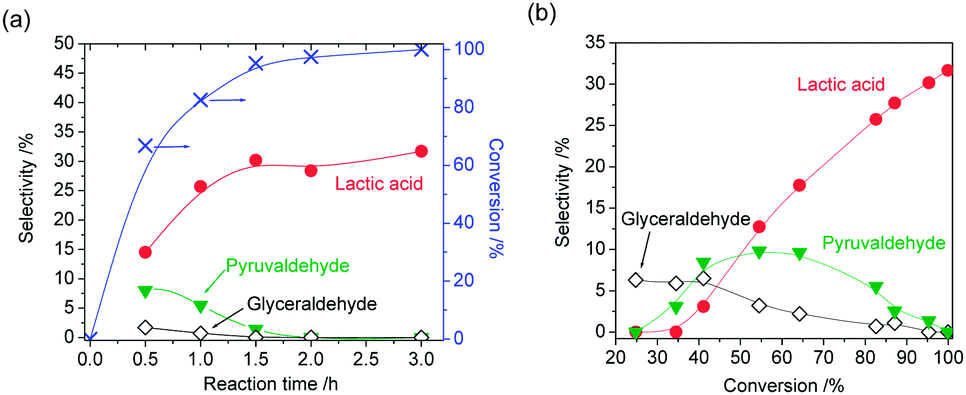 | ||
| Fig. 7 (a) Time-course of product distribution (Reaction conditions: dihydroxyacetone dimer (0.28 mmol), boehmite (50 mg), water (3 mL), 433 K) and (b) dependence of product selectivity on dihydroxyacetone conversion (Reaction conditions: dihydroxyacetone dimer (0.28 mmol), boehmite (50 mg), water (3 mL), 353–433 K, 1–3 h). | ||
Moreover, transformation of hexoses (glucose and fructose) over boehmite in water was investigated. The results are shown in Fig. 8. At the initial stage of the reaction of glucose, fructose was formed with high selectivity (59%) (Fig. 8(a)). With increase of reaction time the selectivity of fructose gradually decreased and the selectivity of lactic acid increased. Interestingly, 5-hydroxymethylfurfural (HMF) was also simultaneously formed. The selectivity of fructose, lactic acid and HMF at 92% conversion was 4%, 26% and 17%, respectively. Other products could be humins because colour of boehmite was changed during the reaction. Similarly, both lactic acid and HMF were simultaneously formed when fructose was used as a reactant (Fig. 8(b)). The selectivity of lactic and HMF at 89% conversion was 16% and 20%, respectively. It is widely accepted that Lewis acid can catalyse not only isomerization of glucose into fructose but also retro-aldol reaction of hexoses into trioses.3 Lewis acid is also effective for the formation of HMF.7 Therefore, boehmite functioned as solid Lewis acid catalyst in water and gave both HMF and lactic acid from hexoses, simultaneously.
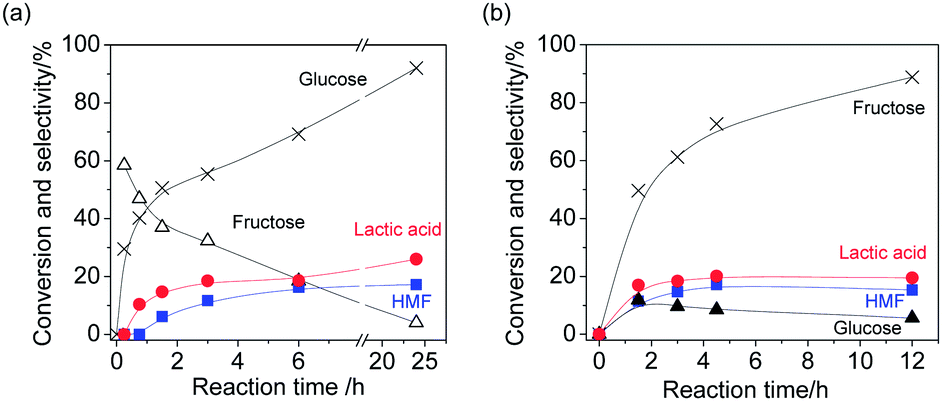 | ||
| Fig. 8 (a) Time-course of product distribution of glucose transformation (Reaction conditions: glucose (0.28 mmol), boehmite (50 mg), water (3 mL), 443 K) and (b) time-course of product distribution of fructose transformation (Reaction conditions: fructose (0.28 mmol), boehmite (50 mg), water (3 mL), 433 K). | ||
Conclusions
Solid Lewis acid property of boehmite was examined by using FTIR and 31P MAS NMR. Even though boehmite has a small amount of weak Lewis acid sites, it exhibited catalytic activity for transformation of triose and hexose in water. Lactic acid was selectively formed from DHA and glyceraldehyde via formation of pyruvaldehyde as an intermediate where 42% yield of lactic acid was obtained from glyceraldehyde. Both lactic acid and HMF were simultaneously formed from glucose and fructose. The selective formation of lactic acid and HMF were ascribed to weak Lewis acid sites of boehmite which catalysed a variety of reactions including isomerization, retro-aldol condensation, dehydration and hydration in water.Acknowledgements
This work was supported by JSPS and NRF under the Japan-Korea Basic Scientific Cooperation Program, and a Grant-in-Aid for Young Scientists (A) (no. 25709077) of JSPS, Japan. A part of this work was conducted at AIST Nanocharacterization Facility.Notes and references
- P. Mäki-Arvela, I. L. Simakova, T. Salmi and D. Y. Murzin, Chem. Rev., 2014, 114, 1909–1971 CrossRef PubMed.
- E. Taarning, S. Saravanamurugan, M. S. Holm, J. M. Xiong, R. M. West and C. H. Christensen, ChemSusChem, 2009, 2, 625–627 CrossRef PubMed.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- R. M. West, M. S. Holm, S. Saravanamurugan, J. Xiong, Z. Beversdorf, E. Taarning and C. H. Christensen, J. Catal., 2010, 269, 122–130 CrossRef CAS PubMed.
- P. Y. Dapsens, C. Mondelli and J. Pérez-Ramirez, ChemSusChem, 2013, 6, 831–839 CrossRef PubMed.
- F. de Clippel, M. Dusselier, R. van Rompaey, P. Vanelderen, J. Dijkmans, E. Makshina, L. Giebeler, S. Oswald, G. V. Baron, J. F. M. Denayer, P. P. Pescarmona, P. A. Jacobs and B. F. Sels, J. Am. Chem. Soc., 2012, 134, 10089–10101 CrossRef PubMed.
- K. Nakajima, Y. Baba, R. Noma, M. Kitano, J. N. Kondo, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2011, 133, 4224–4227 CrossRef PubMed.
- F. Yang, Q. Liu, M. Yue, X. Bai and Y. Du, Chem. Commun., 2011, 47, 4469–4471 RSC.
- K. Nakajima, R. Noma, M. Kitano and M. Hara, J. Mol. Catal. A: Chem., 2014, 388–389, 100–105 CrossRef PubMed.
- S. Dutta, S. De, A. K. Patra, M. Sasidharan, A. Bhaumik and B. Saha, Appl. Catal., A, 2011, 409–410, 133–139 CrossRef PubMed.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef PubMed.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef PubMed.
- R. M. Ravenelle, J. R. Copeland, W.-G. Kim, J. C. Crittenden and C. Sievers, ACS Catal., 2011, 1, 552–561 CrossRef.
- A. Takagaki, K. Iwatani, S. Nishimura and K. Ebitani, Green Chem., 2010, 12, 578–581 RSC.
- C. A. Emieis, J. Catal., 1993, 141, 347–354 CrossRef.
- V. J. Ingram-Jones, R. C. T. Slade, T. W. Davies, J. C. Southern and S. Salvador, J. Mater. Chem., 1996, 6, 73–79 RSC.
- W. H. Chen, H. H. Ko, A. Sakthivel, S. J. Huang, S. H. Liu, A. Y. Lo, T. C. Tsai and S. B. Liu, Catal. Today, 2006, 116, 111–120 CrossRef PubMed.
- Y. Koito, K. Nakajima, R. Hasegawa, H. Kobayashi, M. Kitano and M. Hara, Catal. Today, 2014, 226, 198–203 CrossRef PubMed.
- L. Baltusis, J. S. Frye and G. E. Maciel, J. Am. Chem. Soc., 1986, 108, 7120–7121 CrossRef.
- M. D. Karra, K. J. Sutovich and K. T. Muller, J. Am. Chem. Soc., 2002, 124, 902–903 CrossRef PubMed.
- S. J. Huang, Y. H. Tseng, Y. Mou, S. B. Liu, S. H. Huang, C. P. Lin and J. C. C. Chan, Solid State Nucl. Magn. Reson., 2006, 29, 272–277 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |