
The palladium(II) complex of N,N-diethyl-1-ferrocenyl-3-thiabutanamine: synthesis, solution and solid state structure and catalytic activity in Suzuki–Miyaura reaction†
Ivan Damljanovića,
Dragana Stevanovića,
Anka Pejovića,
Danijela Ilićb,
Marija Živkovića,
Jovana Jovanovića,
Mirjana Vukićevićc,
Goran A. Bogdanovićd,
Niko S. Radulović*e and
Rastko D. Vukićević*a
aDepartment of Chemistry, Faculty of Science, University of Kragujevac, R. Domanovića 12, 34000 Kragujevac, Serbia. E-mail: vuk@kg.ac.rs; Fax: +381-34-34-50-40; Tel: +381-34-30-02-68
bFaculty of Technical Sciences Kosovska Mitrovica, University of Priština, Kneza Miloša 7, 38220 Kosovska Mitrovica, Serbia
cDepartment of Pharmacy, Faculty of Medical Sciences, University of Kragujevac, S. Markovića 69, 34000 Kragujevac, Serbia
dVinča Institute of Nuclear Sciences, University of Belgrade, PO Box 522, 11001 Belgrade, Serbia
eDepartment of Chemistry, Faculty of Science and Mathematics, University of Niš, Višegradska 33, 18000 Niš, Serbia. E-mail: nikoradulovic@yahoo.com; Fax: +381-18-533-014; Tel: +381-62-80-49-210
First published on 29th August 2014
Abstract
In this paper we wish to present the first results on the synthesis of N,N-diethyl-1-ferrocenyl-3-thiabutanamine, its coordination with palladium(II), the complete characterization of the thus obtained complex (including single crystal X-ray analysis for the complex in two polymorphic forms) and screening of its catalytic activity in Suzuki–Miyaura coupling of phenylboronic acid with several aryl bromides. The complex, either purified and then added to the reaction mixture or generated in situ, proved to be an excellent precatalyst in Suzuki–Miyaura coupling. The chemical behavior of the complex in solution was assessed by detailed NMR analyses and cyclic voltammetry measurements which allowed us to draw a number of mechanistic conclusions.
Introduction
Among unnatural compounds, ferrocene and its derivatives have captured particular attention from many chemists due to several unique properties of this metallocene. Utilizing classical methods of organic synthesis (initiated by Woodward's discovery of Friedel–Crafts acylation of ferrocene1), a plethora of ferrocene derivatives have been synthesized until now. These compounds are highly appreciated due to their outstanding stability in both aqueous and non-aqueous media, and have applications in numerous fields, particularly in synthesis and catalysis.2 A multitude of ferrocene-containing ligands are of great interest in coordination chemistry,3 and complexes of transition metals with such ligands are widely used as catalysts in organic synthesis.2 Hence, the synthesis of ferrocene derivatives containing one or more additional heteroatoms (monodentate ligands capable of coordinating with transition metals' cations, or polydentate ligands capable of forming chelates with these cations, respectively) is of high interest for a wide range of chemists. Recently, we reported on the synthesis of several acylferrocenes containing a sulfur atom in their alkyl chains.4 Considering the known reactivity of the carbonyl group, acylferrocenes containing a heteroatom in alkyl chains are much appreciated synthetic starting materials. For example, the synthetic value of such compounds has recently been recognized through the use of sulfonium salts of 1-ferrocenyl-3-thiabutan-1-one and 1-ferrocenyl-4-thiapentan-1-one in cyclopropanation reactions of conjugated enones5 or substitution reactions with diverse nucleophiles.6 The synthesis of compounds containing a new carbon–carbon bond is the particular advantage of the last reaction. Furthermore, the alcohols obtained by the reduction of these compounds can undergo different reactions based on the known stability of carbocations bearing a ferrocenyl group. One of the possible synthetic applications of sulfur-containing acylferrocenes is the synthesis of bidentate ligands (with sulfur as one of the two coordinating heteroatoms) by a formal substitution of the hydroxyl group with certain nucleophiles, e.g. with an amino nitrogen. Such compounds could serve as good phosphine-free ligands for catalysts promoting the formation of new carbon–carbon bonds.Setting up the carbon backbone is one of the basic synthetic tasks and, therefore, the formation of new carbon–carbon bonds lies at the heart of organic synthesis.7 Among many reactions used for this purpose, metal-mediated couplings represent important tools in carbon–carbon bond formation and are widely used in various chemical and pharmaceutical processes. Since its discovery, the Suzuki–Miyaura coupling8 represents one of the most important and widely used reactions in the synthesis of diverse biaryls.8c,9 The most frequently used catalysts for this purpose are palladium complexes with phosphine ligands, among which numerous ferrocene-containing coordination complexes.2,10 The disadvantages of utilizing phosphines as ligands for Pd are numerous, including their sensitivity, high expense, and lack of commercial sources for late generation compounds. Perhaps most importantly, few catalysts exhibit superior activity among broad substrate classes and reaction paradigms. It should also be noted that Pd(PPh3)4 is widely applied in catalysis, but this complex suffers from poor stability upon storage as well as advised handling under nitrogen.11
Results and discussion
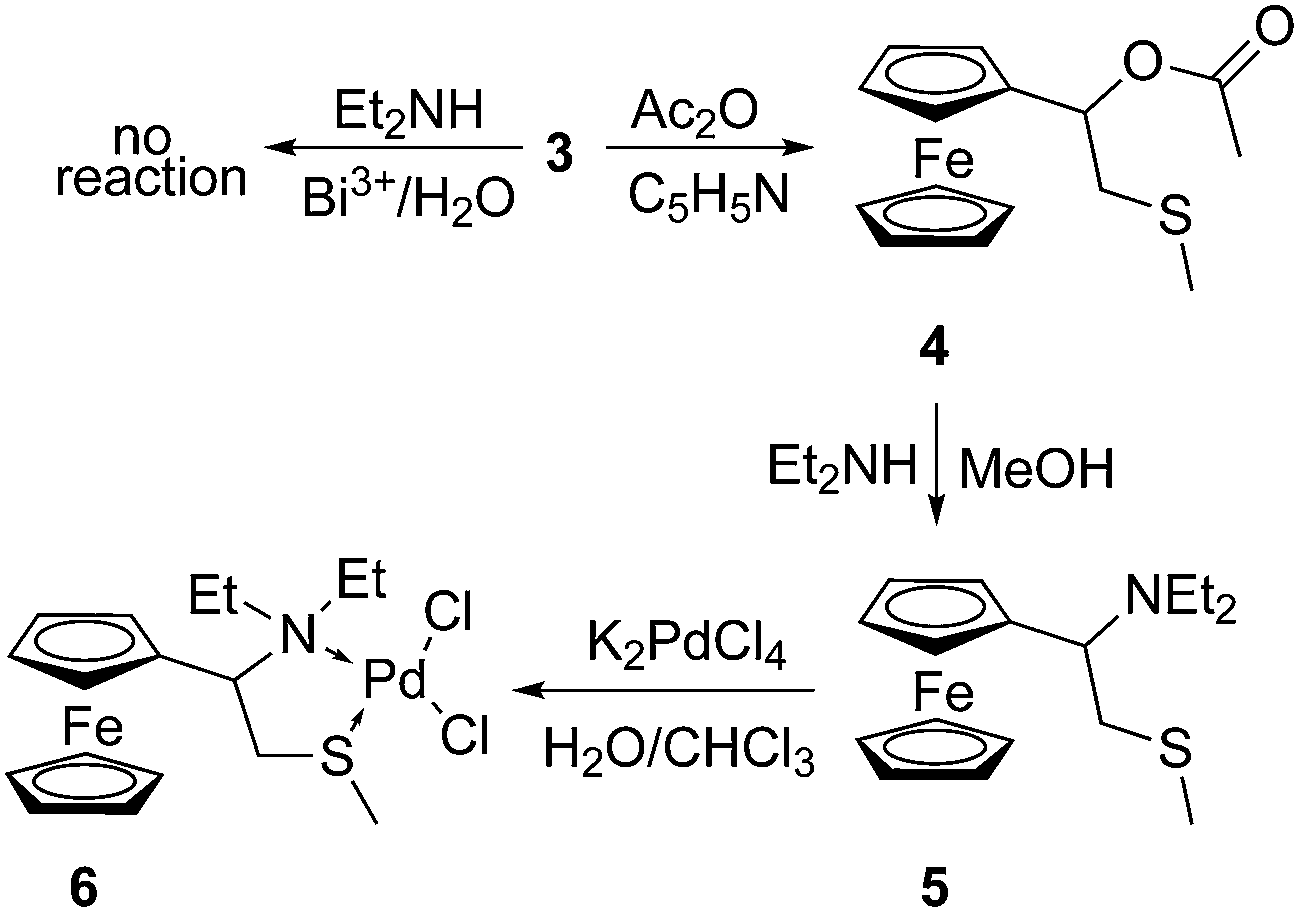
In the present communication we wish to outline the first results on the synthesis of N,N-diethyl-1-ferrocenyl-3-thiabutanamine, its coordination with palladium(II), the complete characterization of the thus obtained complex and screening of its catalytic activity in Suzuki–Miyaura couplings of phenylboronic acid with several aryl bromides.This study started with the synthesis of 1-ferrocenyl-3-thiabutan-1-one (2) by acylation of ferrocene (1) with the in situ generated chloride of S-methylthioglycolic acid (utilizing the procedure developed by us4,12), and the subsequent reduction of this ketone by sodium borohydride in methanol to 1-ferrocenyl-3-thiabutan-1-ol (3)13 (Scheme 1).
 | ||
| Scheme 1 Synthesis of alcohol 3. | ||
The obtained hydroxysulfide 3 was the key intermediate towards the target complex, since the simple substitution of its hydroxyl group with a diethylamino one (the key reaction) should give the target ligand – a bidentate ligand containing an amine nitrogen and a sulfide sulfur atom. Prompted by the recent literature report of a successful direct substitution of the hydroxyl group of 1-ferrocenylethanol with several N-, S- and even C-nucleophiles catalyzed by bismuth(III) nitrate,14 we submitted the alcohol 3 and diethylamine to the conditions described therein (Scheme 2).
 | ||
| Scheme 2 Synthesis of complex 6. | ||
However, in our hands this procedure failed to provide the target product, and we decided to apply the forty-year-old procedure utilized in an indirect substitution of the hydroxyl group from a 1-ferrocenylalkanol with dimethylamine.15 Namely, this method includes the acetylation of an α-ferrocenylalkanol and the subsequent substitution of the acetate from the obtained ester with an amino group. The outstanding stability of the α-ferrocenylcarbocation, i.e. the involvement of the ferrocene core in stabilizing either the intermediate or the transition state, most likely plays the decisive role in directing the reaction into the desired course (alkylation of the nitrogen atom). In this way the hydroxysulfide 3 (via its acetate 4) was converted into the aminosulfide 5 (Scheme 2) in moderate yield (47% based on 3). The structures of the new compounds (ester 4 and aminosulfide 5) were corroborated by spectral data (IR, 1H and 13C NMR).
In the next step the S,N-bidentate ligand 5 was coordinated to palladium(II) ions by reacting this aminosulfide with potassium tetrachloropalladate in a two-phasic solvent system (water/chloroform), giving the complex 6 in 88% yield.
Crystals of complex 6 suitable for X-ray analysis were obtained by slow evaporation of solvents from an ethanol–dichloromethane solution of 6. Compound 6 crystallized with one molecule of ethanol in the asymmetric unit of 6·EtOH (polymorph I). Fig. 1 shows the molecular diagram of this compound with the atom numbering and selected bond lengths and angles. Crystal data and structure refinement are described in ESI.† The ferrocenyl ligand binds to Pd(II) through N1 nitrogen and S1 sulfur atoms, thus confirming a bidentate (N,S) coordination mode of the ligand. The remaining coordination sites of Pd(II) are occupied by two chlorines. All four atoms in the Pd coordination sphere and Pd1 atom are almost ideally coplanar (root-mean-square deviation of the five atoms is only 0.005 Å). However, the square-planar geometry around Pd(II) is rather distorted since the coordination angles significantly deviate from 90°. Thus, N1–Pd1–Cl2 and S1–Pd1–Cl1 are 93.73(8) and 86.77(4)°, respectively. Two Pd–Cl bonds show a difference in bond lengths of almost 0.4 Å.
 | ||
| Fig. 1 The molecular structure of 6 with atom numbering scheme (hydrogen atoms and ethanol solvent molecule are omitted for clarity). Displacement ellipsoids are drawn at the 30% probability level. Other projections of the molecule are given in Fig. S1 (see ESI†). Selected bond lengths (Å) and angles (°): Pd1–N1 = 2.107(3), Pd1–S1 = 2.2350(10), Pd1–Cl1 = 2.3016(11), Pd1–Cl2 = 2.3387(11), N1–C16 = 1.496(5), N1–C14 = 1.504(5), N1–C11 = 1.543(5); N1–Pd1–S1 = 87.92(8), N1–Pd1–Cl1 = 174.67(9), S1–Pd1–Cl1 = 86.77(4), N1–Pd1–Cl2 = 93.73(8), S1–Pd1–Cl2 = 178.34(4), Cl1–Pd1–Cl2 = 91.58(5). | ||
The five-membered Pd1–N1–C11–C12–S1 chelate ring adopts a twisted conformation with the N1–C11–C12–S1 torsion angle of 51.3(4)°. Both ethyl groups bonded to N1 atom have very similar space directionality and an almost orthogonal orientation in relation to the Pd(II) coordination plane. Dihedral angle between the N1–C14–C15 and N1–C16–C17 mean planes is 12.0(5)°. The N1–C11 bond has bond length which is about 0.4 Å longer than for the N1–C14 and N1–C16 bonds. Bond lengths and angles of the ferrocenyl unit agree with those expected for monosubstituted ferrocene derivatives. The two cyclopentadienyl rings (Cp) are planar and nearly parallel. Dihedral angle between two Cp rings is 2.5(5)°. The Cp rings significantly deviate from an eclipsed conformation (see Fig. S1 in the ESI†). The C1–Cg1–Cg2–C6 torsion angle is 21.3° (Cg1 and Cg2 are centroids of the corresponding Cp rings).
In the crystal, molecules of 6 form very interesting dimers. The first centrosymmetric dimer (Fig. S2†) possesses two Pd⋯S intermolecular contacts with the Pd1⋯S1i distance (3.76 Å) close to the sum of van der Waals radii. The Pd1⋯S1i vector [symmetry code: (i) −x + 1, −y + 1, −z + 2] agrees very well with the potential directionality of the lone pair at the sulfur atom. Also the location of S1i corresponds to the apical position in the coordination sphere of Pd1. This probably stabilizing intermolecular interaction within the dimer is also supplemented by several C–H⋯Cl, C–H⋯O and O–H⋯Cl intermolecular interactions (Fig. S3†). Some of these interactions are formed through two EtOH molecules. Practically all significant interatomic interactions are located within this dimer.
Another interesting dimer is formed between two ferrocene units placed in parallel orientation (Fig. S4†). Recently it has been reported16 that almost 60% of crystal structures containing monosubstituted ferrocene (Fc) form a Fc–Fc dimer as a common building block. It has been shown that two Fc units within the dimer exhibit an excellent electrostatic complementarity in a very large contact surface of about 6 × 3 Å2. Such a dimer also exists in the crystal structure of 6·EtOH (Fig. S4†). In that way the crystal packing of 6·EtOH is stabilized by the formation of two types of dimers as dominant building blocks. One formed by Pd coordination sphere and another one formed by the ferrocene unit.
1H and 13C NMR spectra of complex 6 were noticeably different from that of the uncoordinated ligand; addition of the free ligand resulted in another set of proton (Table 1) and carbon-13 (Table 2) signals with the expected chemical shifts. The palladation of the ligand resulted in a mean 1H and 13C deshielding (mean ΔH: +0.80 ppm, mean ΔC: +2.7 ppm) as deduced from the unassigned NMR data. The observed downfield shift could be explained by the expected electron-withdrawing effect of palladium(II), i.e. the overall electron density should be more toward the metal halide which in effect would make the coordinating atoms electron-deficient compared to the free ligands.
| Hydrogen | 5 | Cis-6a | Trans-6a |
|---|---|---|---|
| a NMR spectra of 6·EtOH were identical to those reported in this table except for the additional presence of a triplet (1.24 ppm, J = 7.0 Hz, 3H, CH3) and quartet (3.71 ppm, J = 7.0 Hz, 2H, CH2) pair of coupled signals and an exchangeable broad signal (1.21 ppm, brs, 1H, OH) pertaining to ethanol.b On the same side of the chelate ring as the ferrocenyl group. | |||
| 1 | 3.87 (dd, J = 9.9, 4.4 Hz, 1H) | ∼4.22 (overlapped, 1H) | 4.73 (dd, J = 13.4, 4.3 Hz, 1H) |
| 2 CHaHbS | 3.07 (dd, J = 12.8, 4.4 Hz, 1H) | 3.67 (dd, J = 12.6, 3.4 Hz, 1H) | 3.67 (dd, J = 15.1, 13.4 Hz, 1H) |
| CHaHbS | 3.01 (dd, J = 12.8, 9.9 Hz, 1H) | 3.35 (dd, J = 13.8, 12.6 Hz, 1H)b | 3.21 (dd, J = 15.1, 4.3 Hz, 1H)b |
| CH3S | 2.25 (s, 3H) | 2.75 (s, 3H)b | 2.80 (s, 3H) |
| N(CH2CH3)2 | 2.48 (dq, J = 12.9, 7.3 Hz, 2H, N(CHaHbCH3)2) | 3.49 (dq, J = 13.2, 7.0 Hz, 1H, CHaHbCH3) | 3.59 (dq, J = 13.2, 7.2 Hz, 1H, CHaHbCH3) |
| 2.94 (dq, J = 13.2, 7.5 Hz, 1H, CHaHbCH3)b | 2.63 (dq, J = 13.1, 7.3 Hz, 1H, CHaHbCH3)b | ||
| 2.06 (dq, J = 12.9, 7.0 Hz, 2H, N(CHaHbCH3)2) | 2.30 (dq, J = 13.2, 6.7 Hz, 1H, CHaHbCH3) | 2.15 (dq, J = 13.2, 6.4 Hz, 1H, CHaHbCH3) | |
| 2.43 (dq, J = 13.2, 6.9 Hz, 1H, CHaHbCH3)b | 2.52 (dq, J = 13.1, 7.3 Hz, 1H, CHaHbCH3)b | ||
| N(CH2CH3)2 | 1.01 (brt, J = 7.1 Hz, 6H) | 1.87 (brt, J = 6.8 Hz, 3H) | 1.88 (brt, J = 6.8 Hz, 3H) |
| 1.76 (brt, J = 7.2 Hz, 3H)b | 1.67 (t, J = 7.3 Hz, 3H)b | ||
| C5H4 | 4.04 (dt, J = 2.3, 1.3 Hz, 1H), 4.14–4.09 (overlapped, 3H) | 4.41–4.32 (Overlapped, 4H), 4.31–4.25 (overlapped, 3H), 4.24–4.18 (overlapped, 1H) | |
| C5H5 | 4.12 (s, 5H) | 4.19 (s, 5H) | 4.16 (s, 5H) |
| Carbon | 5 | Cis-6a | Trans-6a |
|---|---|---|---|
| a NMR spectra of 6 EtOH were identical to those reported in this table except for the additional presence of two signals at 18.5 ppm (CH3) and 58.5 ppm (CH2) corresponding to ethanol.b On the same side of the chelate ring as the ferrocenyl group. | |||
| 1 | 58.8 | 66.7 | 67.4 |
| 2 | 37.6 | 43.5 | 40.0 |
| CH3S | 16.8 | 24.0b | 22.0 |
| N(CH2CH3)2 | 43.6 | 53.3, 52.9b | 51.7, 52.1b |
| N(CH2CH3)2 | 14.1 | 13.9, 15.3b | 14.2, 15.2b |
| C5H4 | 68.6 | 67.5 | 70.1 |
| (C-2′-C-5′) | 67.2(3), 67.2(0), 66.9 | 68.2, 69.3(7), 69.6(4) | 69.9, 69.4(1), 69.4(1) |
| (C-1′) | 86.4 | 76.2 | 75.7 |
| C5H5 | 68.6 | 69.6(4) | 69.5(7) |
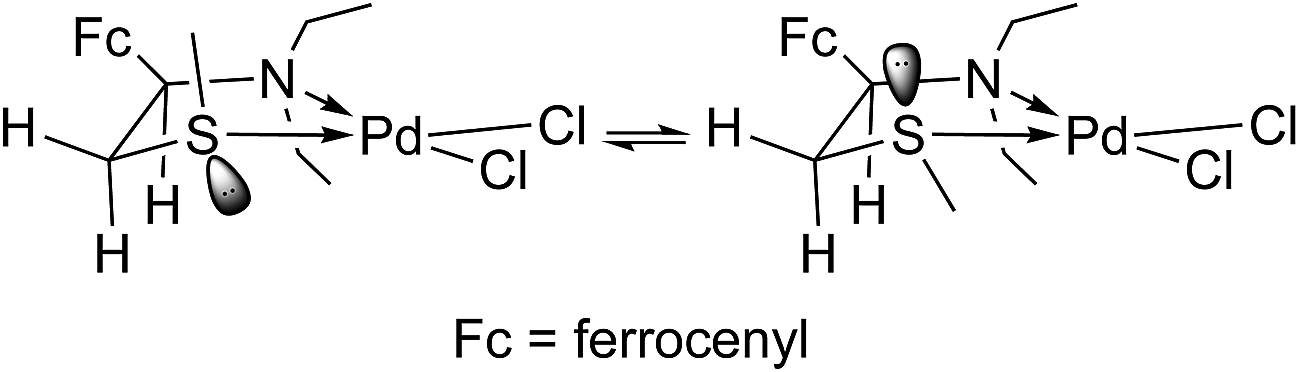
As depicted in Fig. 1, complex 6 in solid state has a cis-configuration (the methyl group at the sulfur atom and the ferrocenyl group are on the same side of the PdSCCN ring). However, its NMR spectra in CDCl3 always corresponded to a two component mixture of constant composition (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). After a detailed analysis of both 1D- (1H, 13C and DEPT) and 2D-(1H–1H–COSY, NOESY, HSQC and HMBC) NMR spectra, and a series of selective homodecoupling experiments, all of the observed signals were successfully assigned to the two diastereomeric 6, differing in the relative stereochemistry around the chelate ring. The signals of methylthio protons of both diastereomers at 2.75 and 2.80 ppm (cis and trans, respectively) were the entry points in 2D spectra that corroborated the expected connectivity of the molecules. Crucial pieces of information that enabled an almost complete assignation of the trans-isomer (Table 1) came from the NOESY spectrum. More precisely, the nOe crosspeak between SCH3 group protons and H-1 proton, both of which simultaneously interacted with H-2a, suggested a spatial proximity of the mentioned hydrogens. The values of the coupling constants of H-1 and H-2a or H-2b (cis-6 J3(H-1-H-2a) = 4.3 Hz, J3(H-1-H-2b) = 13.4 Hz, trans-6 J3(H-1-H-2a) = 12.8 Hz) corroborate this stereochemical relationship. The coordination to Pd produced a new chiral center (S atom) and caused a differentiation of the protons from the ethyl groups (both CH2 and CH3 now gave rise to a myriad of signals – 4 different methyl group triplets and 8 doublets of quartets originating from the diastereotopic CH2 protons), and this also clearly demonstrates the existence of the cis- and trans-6. Due to strong mutual overlap the only unresolved protons left in the spectra (in the sense to which of the two isomers they belong to) were those of the ferrocene moiety.
:1). After a detailed analysis of both 1D- (1H, 13C and DEPT) and 2D-(1H–1H–COSY, NOESY, HSQC and HMBC) NMR spectra, and a series of selective homodecoupling experiments, all of the observed signals were successfully assigned to the two diastereomeric 6, differing in the relative stereochemistry around the chelate ring. The signals of methylthio protons of both diastereomers at 2.75 and 2.80 ppm (cis and trans, respectively) were the entry points in 2D spectra that corroborated the expected connectivity of the molecules. Crucial pieces of information that enabled an almost complete assignation of the trans-isomer (Table 1) came from the NOESY spectrum. More precisely, the nOe crosspeak between SCH3 group protons and H-1 proton, both of which simultaneously interacted with H-2a, suggested a spatial proximity of the mentioned hydrogens. The values of the coupling constants of H-1 and H-2a or H-2b (cis-6 J3(H-1-H-2a) = 4.3 Hz, J3(H-1-H-2b) = 13.4 Hz, trans-6 J3(H-1-H-2a) = 12.8 Hz) corroborate this stereochemical relationship. The coordination to Pd produced a new chiral center (S atom) and caused a differentiation of the protons from the ethyl groups (both CH2 and CH3 now gave rise to a myriad of signals – 4 different methyl group triplets and 8 doublets of quartets originating from the diastereotopic CH2 protons), and this also clearly demonstrates the existence of the cis- and trans-6. Due to strong mutual overlap the only unresolved protons left in the spectra (in the sense to which of the two isomers they belong to) were those of the ferrocene moiety.
The existence of two diastereoisomers of 6 (of equal thermodynamic stability) in solution at room temperature can be interpreted in two ways: they interconvert rapidly in solution and one of the isomers is less soluble than the other, or the solid state is also consisted of crystals of both cis- and trans-6. Since complex 6 displays sharp signals of both isomers in the NMR spectra, the rate of interconversion could be either very slow or could be in the domain of medium slow (up to about a minute) exchanges, yielding sharp separate spectra but also exchange peaks in the NOESY spectrum. Since the later were observed in our spectra (for example the interaction of H-1 at 4.73 and 4.22 ppm from trans- and cis-6, respectively), we could conclude that the equilibrium shown in Scheme 3 is medium slow in CDCl3 at 25 °C.
 | ||
| Scheme 3 Equilibrium between diastereoisomers of 6 in solution. | ||
Apparently, the cis-/trans-diastereoisomers of 6 interconvert relatively easily in the solution, whereas it seems that only the cis-isomer crystallizes out of solution. In order to acquire an additional evidence for this, complex 6 was recrystallized from several other solvents and solvent mixtures. Only recrystallization from methanol gave another crystal polymorph(II) with non-solvated Pd complex molecules (see ESI†). Although these crystals were of poor crystallographic quality (racemic twinning) the single-crystal X-ray analysis confirmed undoubtedly the cis-configuration of 6 in this crystal polymorph (see Fig. S5†) as it was found in the crystal structure of 6·EtOH (Fig. 1). The main conformational difference of the Pd complex in two polymorphs is the mutual orientation of the ethyl groups bonded to the N1 atom (Fig. S7†). Since thiomorpholine and Pd(II) form a PdL2X2 complex with proven N-coordination,17 the equilibrium between the two diastereoisomers in solution proceeds most likely through the breaking of the S–Pd bond, as depicted in Scheme 3.
Redox properties of ligand 5 and complex 6 were evaluated by cyclic voltammetry in a 0.1 M dichloromethane solution of tetrabutylammonium perchlorate (at a glassy carbon disc working electrode). On the basis of preliminary measurements we have chosen a −1.500/+1.500 V potential window and the scan rate ν = 0.2 V s−1. As depicted in Fig. 2 (the first and second scan, curves a and b, respectively), ligand 5 exhibits four oxidation (O1–4) and four reduction waves (R1–4). When the potential swept up to 0.800 V, only O2 and R2 were observed (curve c), and we attributed these waves to the ferrocene unit. The redox potential of this couple is the same as for the unsubstituted ferrocene under the same conditions (curve d).
 | ||
| Fig. 2 Cyclic voltammetry (ν = 0.2 V s−1) of 5 (5 × 10−4 M) in 0.1 M Bu4NClO4 at a glassy carbon disc electrode (2 mm diameter). | ||
The true nature of processes responsible for waves O1, O3, O4, R1, R3 and R4, for which the lone electron pairs of heteroatoms are responsible, exceeds the scope of this paper. The lack of these waves in the voltammogram of complex 6 (Fig. 3), in which these electrons are engaged in the formation of Pd–N and Pd–S coordinative bonds, confirms this claim. As depicted in Fig. 3 (curve a), complex 6 exhibits two oxidation waves on the forward potential sweep (O1 at 0.533 and O2 at 0.763 V, respectively) and two reduction waves on the back potential sweep (R1 at 0.625 and R2 at 0.994 V, respectively). We attributed the O2/R2 response of the working electrode (curve a) to the ferrocene unit, which is slightly shifted to higher oxidation potentials in comparison with the ferrocene/ferrocenium couple (curve b). This could be rationalized by the increase of electronegativity of the coordinated nitrogen (as much as to prevent its oxidation in the chosen potential window), which changes the electronic properties of the carbon atom next to the ferrocene unit, making it more electron-withdrawing.
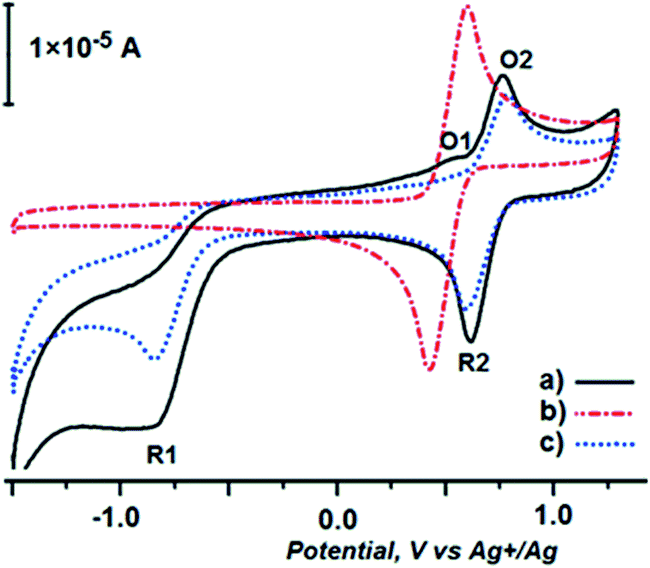 | ||
| Fig. 3 Cyclic voltammetry (ν = 0.2 V s−1) of 6 (5 × 10−4 M) in 0.1 M Bu4NClO4 at a glassy carbon disc electrode (2 mm diameter). | ||
The second electrochemically sensitive position of complex 6, the coordinated palladium ion, under the applied conditions, exhibits one oxidation and one intensive reduction peak (O1 and R1, respectively). These waves reflect the reduction of coordinated PdII and subsequent oxidation of the obtained Pd0 species. Following the idea described in the literature for Pd(PPh3)2Cl2,18 in the next experiment, cyclovoltammetry of complex 6 was conducted in the presence of iodobenzene. O1 wave disappeared (curve c), since Pd0 species underwent oxidative addition giving a pentacoordinated complex (see Scheme 4). This complex should undergo oxidation at higher potentials,18 but in the present case the corresponding wave cannot be seen due to overlapping with the O2 wave.
 | ||
| Scheme 4 Reduction of 6 at the working electrode and trapping of the obtained Pd0 species. | ||
The catalytic utility of complex 6 in the Suzuki–Miyaura reaction was evaluated in the following manner: isolated crystals of this compound were used as the catalyst (1 mol%) in an overnight reaction of phenylboronic acid (7) with bromobenzene (8a) in a 4:1 (v/v) DMF–water mixture (Scheme 5) in the presence of K2CO3. The experiment conducted at room temperature resulted in the synthesis of biphenyl 9a in 45% yield, but the reaction run at 70 °C gave this compound in almost quantitative yield (Table 3, entries 1 and 2, respectively). To make this procedure more attractive from the synthetic point of view, i.e. more suitable for practical use, we repeated the experiment without the isolation and purification of the catalyst. Namely, to the mixture of equimolar amounts (0.01 mmol) of ligand 5 and palladium(II) chloride (or acetate), stirred for two hours in the same solvent at 70 °C (bath temperature), bromobenzene (1 mmol), phenylboronic acid (1.2 mmol) and K2CO3 were added, and stirring was continued overnight. The reaction where the catalyst was generated in situ was as successful as the previous one (entry 3, Table 3).
 | ||
| Scheme 5 The Suzuki–Miyaura couplings of phenylboronic acid with organic halides catalyzed by complex 6. | ||
| Entry | Aryl halide | Product | Methoda | Yieldb (%) |
|---|---|---|---|---|
| a Method A: Isolated, monosolvated complex 6 was used as the catalyst at room temperature; Method B: isolated, monosolvated complex 6 was used as the catalyst at 70 °C (bath temperature); Method C: in situ formed complex 6 was used as the catalyst at 70 °C (bath temperature).b Isolated yield, based on the starting aryl halide. | ||||
| 1 | 8a | 9a | A | 45 |
| 2 | 8a | 9a | B | 99 |
| 3 | 8a | 9a | C | 99 |
| 4 | 8b | 9b | C | 99 |
| 5 | 8c | 9c | C | 99 |
| 6 | 8d | 9d | C | 94 |
| 7 | 8e | 9e | C | 87 |
| 8 | 8f | 9f | C | 98 |
| 9 | 8g | 9g | C | 96 |
| 10 | 8h | 9h | C | 87 |
| 11 | 8i | 9i | C | 98 |
| 12 | 8j | 9j | C | 83 |
| 13 | 8k | 9k | C | 50 |
In order to evaluate the generality of this reaction, we employed nine additional bromoarenes as substrates in this reaction (8b–j, Scheme 5), but using the in situ generated catalyst only. As the data listed in Table 3 show (entries 4–12), dichlorido-(N,N-diethyl-1-ferrocenyl-3-thiabutanamine)palladium(II) (6) can be used as an efficient precatalyst in the Suzuki–Miyaura couplings of aryl bromides with phenylboronic acid. On the other hand, p-nitrochlorobenzene (8k), the only aryl chloride used in this study, under the same reaction conditions, gave the corresponding biphenyl in considerably lower yield (entry 13, Table 3).
The catalytic activity of palladium chelates is known, and recently several complexes of this metal with N- or P-chelating ligands were examined as catalysts in the Suzuki–Miyaura reaction by both computational and experimental studies.19 The proposed mechanism does not include the chelate ring opening; applying these statements, the mechanism of the catalytic action of complex 6 might be presented as depicted in Scheme 6, pathway A. However, in the light of the abovementioned considerations regarding the easiness by which cis- and trans-6 interconvert (i.e. the existence of a labile ligand –S-atom of compound 5), the mechanism similar to that accepted for palladium complexes with monodentate ligands,18 i.e. the mechanism including the chelate ring opening, should not be disregarded (Scheme 6, pathway B).
 | ||
| Scheme 6 The Suzuki–Miyaura couplings of phenylboronic acid with organic halides catalyzed by complex 6. | ||
Unlike traditional palladium phosphine and NHC (nucleophilic heterocyclic carbene) catalysts, compound 6 can be stored outside an inert atmosphere. The precatalyst may be weighed out on bench utilizing normal methods and can even be subjected to a water workup without observable decomposition by 1H NMR. This Pd(II) complex becomes active in situ through reduction to the Pd(0) active catalyst—thus it can be considered a ligand stabilized Pd(PPh3)4 alternative, minus the handling deficiencies.
Conclusions
In summary, we described herein the synthesis of a new ferrocene-containing chelate – dichlorido-(N,N-diethyl-1-ferrocenyl-3-thiabutanamine)palladium(II) monosolvate (6) – which was fully characterized by spectral data and single crystal X-ray structure analysis. It has been shown that this complex is an efficient catalyst for Suzuki–Miyaura couplings of phenylboronic acid with aryl bromides. It also turned out that the in situ generated complex is of the same catalytic activity as the isolated one.Experimental section
General remarks
All chemicals were commercially available and used as received, except that the solvents were purified by distillation. Ultrasonic cleaner Elmasonic S 10 (Elma, Germany), 30 W was used for the ultrasonically supported synthesis. Chromatographic separations were carried out using silica gel 60 (Merck, 230–400 mesh ASTM), whereas silica gel 60 on Al plates, layer thickness 0.2 mm (Merck) was used for TLC. Melting points (uncorrected) were determined on a Mel-Temp capillary melting points apparatus, model 1001. The 1H and 13C{1H} NMR spectra of the samples in CDCl3 were recorded at room temperature on a Varian Gemini (200 MHz) and a Bruker Avance III 400 MHz (1H at 400 MHz, 13C at 101 MHz) NMR spectrometers. Chemical shifts are expressed in δ (ppm), relative to the residual solvent protons or 13CDCl3 as internal standards (CHCl3: 7.26 ppm for 1H and 77 ppm for 13C). Standard pulse sequences were used for 2D spectra (1H–1H COSY, NOESY, HSQC and HMBC). IR measurements were carried out with a Perkin-Elmer FTIR 31725-X spectrophotometer. Microanalysis of carbon and hydrogen were carried out with a Carlo Erba 1106 microanalyser; their results agreed favorably with the calculated values.Synthetic procedures
:1). The solvent was left to slowly evaporate (several days) which gave 487.5 mg (0.88 mmol) of dark red crystals of 6·EtOH (88%). m.p. 163 °C; IR (KBr): ν = 3468, 3082, 2964, 2923, 2875, 1631, 1452, 1416, 1043, 1032 cm−1; for NMR data see Tables 1 and 2. Anal. calc. for 6·EtOH = C19H31Cl2FeNPdS (FW = 554.69): C, 41.14; H, 5.63; N, 2.53. Found: C, 41.08; H, 5.56; N, 2.57%.:1, v/v) and placed into a refrigerator. After three days the solvent was decanted and the obtained crystals of unsolvated 6 (m.p. 163 °C) dried on air. IR (KBr): ν = 3082, 2998, 2956, 2922, 2875, 1639, 1452, 1416, 1230, 1106, 1043, 1032 cm−1; for NMR data see Tables 1 and 2. Anal. calc. for 6 = C17H25Cl2FeNPdS (FW = 508.62): C, 40.14; H, 4.95; N, 2.75. Found: C, 40.21; H, 5.02; N, 2.79%.General procedures for Suzuki–Miyaura reaction
X-ray crystal structure analysis of 6
Acknowledgements
We thank to the Ministry of Education, Science and Technological Development of the Republic of Serbia for financial support (Grant 172034).Notes and references
- R. B. Woodward, M. Rosenblum and M. Whiting, J. Org. Chem., 1952, 74, 3458–3459 CAS.
- (a) Ferrocenes: Homogenous Catalysis, Organic Synthesis, Materials Science, ed. A. Togni and T. Hayashi, VCH, Weinheim, Germany, 1995 Search PubMed; (b) Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, Wiley, Chichester, U.K., 2008 Search PubMed.
- (a) W. R. Cullen and J. D. Woollins, Coord. Chem. Rev., 1981, 39, 1–30 CrossRef CAS; (b) K. Osakada, T. Sakano, M. Horie and A. Suzaki, Coord. Chem. Rev., 2006, 250, 1012–1022 CrossRef CAS PubMed.
- (a) R. D. Vukićević, D. Ilić, Z. Ratković and M. Vukićević, Chem. Mon., 2001, 132, 625–631 CrossRef; (b) D. Ilić, I. Damljanović, D. Stevanović, M. D. Vukićević, N. Radulović, V. Kahlenberg, G. Laus and R. D. Vukićević, Polyhedron, 2010, 29, 1863–1869 CrossRef PubMed; (c) Z. Ratković, S. B. Novaković, G. A. Bogdanović, D. Šegan and R. D. Vukićević, Polyhedron, 2010, 29, 2311–2317 CrossRef PubMed.
- D. Ilić, I. Damljanović, M. Vukićević, V. Kahlenberg, G. Laus, N. Radulović and R. D. Vukićević, Tetrahedron Lett., 2012, 53, 6018–6021 CrossRef PubMed.
- A. Pejović, I. Damljanović, D. Stevanović, D. Ilić, M. Vukićević, G. Bogdanović and R. D. Vukićević, Tetrahedron Lett., 2013, 54, 4776–4780 CrossRef PubMed.
- E. J. Corey and X. M. Cheng, The Logic of Chemical Synthesis, John Wiley & Sons, New York, 1989 Search PubMed.
- (a) N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866–867 RSC; (b) N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef; (c) N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513–519 CrossRef CAS; (d) A. Suzuki, in Metal-Catalyzed Cross-Coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, New York, 1998, pp. 49–97 Search PubMed.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS; (c) E. J.-G. Anctil and V. Snieckus, J. Organomet. Chem., 2002, 653, 150–160 CrossRef CAS; (d) S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58, 9633–9695 CrossRef CAS.
- T. J. Colacot, Platinum Met. Rev., 2001, 45, 22–30 CAS.
- Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, John Wiley, New York, 2012 Search PubMed.
- (a) R. D. Vukićević, M. Vukićević, Z. Ratković and S. Konstantinović, Synlett, 1998, 1329–1330 CrossRef PubMed; (b) M. D. Vukićević, Z. R. Ratković, A. T. Teodorović, G. S. Stojanović and R. D. Vukićević, Tetrahedron, 2002, 58, 9001–9006 CrossRef.
- D. Ilić, I. Damljanović, D. Stevanović, M. Vukićević, P. Blagojević, N. Radulović and R. D. Vukićević, Chem. Biodiversity, 2012, 9, 2236–2253 Search PubMed.
- R. Jiang, C.-X. Yuan, X.-P. Xu and S.-J. Ji, Appl. Organomet. Chem., 2012, 26, 62–66 CrossRef CAS PubMed.
- G. W. Gokel and I. K. J. Ugi, J. Chem. Educ., 1972, 49, 294–296 CrossRef CAS.
- G. A. Bogdanovic and S. B. Novakovic, CrystEngComm, 2011, 13, 6930–6932 RSC.
- S. A. Clifford, V. Tiwow, A. Gendron, M. Maeder, M. Rossignoli, G. A. Lawrance, P. Turner, A. J. Blake and M. Schröder, Aust. J. Chem., 2009, 62, 1196–1206 CrossRef CAS.
- C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314–321 CrossRef CAS PubMed.
- Y.-L. Huang, C.-M. Weng and F.-E. Hong, Chem.–Eur. J., 2008, 14, 4426–4434 CrossRef CAS PubMed.
- E. Alacid and C. Nájera, Org. Lett., 2008, 10, 5011–5014 CrossRef CAS PubMed.
- L. Bo, C. Fu and S. Ma, Tetrahedron Lett., 2010, 51, 1284–1286 CrossRef PubMed.
- W.-J. Zhou, K.-H. Wang and J.-X. Wang, Adv. Synth. Catal., 2009, 351, 1378–1382 CrossRef CAS PubMed.
- L. Bai and J.-X. Wang, Adv. Synth. Catal., 2008, 350, 315–320 CrossRef CAS PubMed.
- P. D. Stevens, J. Fan, H. M. R. Gardimalla, M. Yen and Y. Gao, Org. Lett., 2005, 7, 2085–2088 CrossRef CAS PubMed.
- Agilent, CrysAlis PRO, Agilent Technologies, Yarnton, Oxfordshire, England, 2013 Search PubMed.
- M. C. Burla, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2003, 36, 1103 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- M. Nardelli, J. Appl. Crystallogr., 1995, 28, 659 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data, CIF files and copies of 1H and 13C NMR spectra for all newly synthesized compounds, as well as X-ray crystallographic data for 6. CCDC 1016148 and 1016149. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra08140d |
| This journal is © The Royal Society of Chemistry 2014 |