
Click synthesis of graphene/poly(N-(2-hydroxypropyl) methacrylamide) nanocomposite via “grafting-onto” strategy at ambient temperature
Abstract
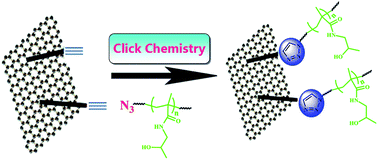
So far, much work has been performed to modify graphene by physical or chemical methods, aiming to improve its solubility in solvents for its further application. Herein, we reported a new graphene/polymer nanocomposite, a graphene/poly(N-(2-hydroxypropyl) methacrylamide) (PHPMA) nanocomposite (G-PHPMA), by combining reversible addition–fragmentation chain transfer (RAFT) polymerization and click chemistry under mild conditions via the “grafting-onto” strategy. The as-prepared azido-terminated PHPMA homopolymer via RAFT polymerization was grafted onto yne-containing graphene through click chemistry at room temperature using CuBr/PMDETA as the catalytic system. FT-IR, XRD, Raman, element analysis, TGA, AFM, and TEM measurements were used to confirm the covalent linkage between PHPMA chains and graphene. The resulting G-PHPMA nanocomposite showed excellent dispersibility in organic solvents and aqueous media and it could quickly enter into SMMC-7721 and SH-SY5Y cells.
- This article is part of the themed collection: A Decade of Progress in Click Reactions Based on CuAAC
 Please wait while we load your content...
Please wait while we load your content...

