
One-pot transesterification and esterification of waste cooking oil via ethanolysis using Sr:Zr mixed oxide as solid catalyst†
Navjot Kaur and
Amjad Ali*
School of Chemistry and Biochemistry, Thapar University, Patiala-147004, India. E-mail: amjadali@thapar.edu; amjad_2kin@yahoo.com; Fax: +91-175-2364498; Fax: +91-175-2393005; Tel: +91-175-2393832
First published on 27th August 2014
Abstract
Mixed oxides of Sr and Zr were prepared by the co-precipitation method and examined as heterogeneous catalysts for the one-pot esterification and transesterification of waste cooking oil with ethanol for the production of fatty acid ethyl esters (FAEE). The mixed oxide of Sr:Zr with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 atomic ratio calcined at 650 °C showed optimum activity among the prepared catalysts. The catalyst possesses both acidic and basic sites, and hence was able to perform simultaneous esterification and transesterification of free fatty acid containing vegetable oils. The transesterification activity was found to be a function of basic sites of the catalyst which in turn depends on calcination temperature and Sr:Zr atomic ratio. A pseudo-first-order kinetic equation was applied to evaluate the kinetics of the 2Sr:Zr-650 catalyzed ethanolysis of waste cottonseed oil and the activation energy (Ea) for the reaction was observed as 48.17 kJ mol−1. The thermodynamic activation parameters of the reaction were evaluated from the Eyring-Polanyi equation and the values of ΔG‡, ΔH‡ and ΔS‡ were found to be 88.23 kJ mol−1, 45.97 kJ mol−1 and −121.37 J mol−1 K−1, respectively. These values suggest that the 2Sr:Zr-650 catalyzed reaction is endothermic, unspontaneous and follows the associative mechanism. The catalyst was recovered by simple filtration from the reaction mixture and reused in four cycles without any significant loss in activity as well as metal leaching from the catalyst in reaction mixture.
:1 atomic ratio calcined at 650 °C showed optimum activity among the prepared catalysts. The catalyst possesses both acidic and basic sites, and hence was able to perform simultaneous esterification and transesterification of free fatty acid containing vegetable oils. The transesterification activity was found to be a function of basic sites of the catalyst which in turn depends on calcination temperature and Sr:Zr atomic ratio. A pseudo-first-order kinetic equation was applied to evaluate the kinetics of the 2Sr:Zr-650 catalyzed ethanolysis of waste cottonseed oil and the activation energy (Ea) for the reaction was observed as 48.17 kJ mol−1. The thermodynamic activation parameters of the reaction were evaluated from the Eyring-Polanyi equation and the values of ΔG‡, ΔH‡ and ΔS‡ were found to be 88.23 kJ mol−1, 45.97 kJ mol−1 and −121.37 J mol−1 K−1, respectively. These values suggest that the 2Sr:Zr-650 catalyzed reaction is endothermic, unspontaneous and follows the associative mechanism. The catalyst was recovered by simple filtration from the reaction mixture and reused in four cycles without any significant loss in activity as well as metal leaching from the catalyst in reaction mixture.
1. Introduction
The use of biodiesel as a substitute for fossil fuels is becoming gradually more popular nowadays due to global energy crisis and environmental reasons. Biodiesel (BD), defined as fatty acid alkyl esters (FAAE), being renewable, biodegradable and non-toxic, has become an eco-friendly substitute for fossil fuel based diesel fuel.1 Additionally, BD burning causes lesser emissions of SOx, CO, unburnt hydrocarbons and particulate matter, making it an eco-friendly fuel. Biodiesel is produced conventionally by the transesterification of edible or non-edible oils and animal fats with short chain alcohols in the presence of homogeneous acid or base catalyst.2,3 Due to limited supply, application of the edible vegetable oil is prohibited for BD production in India. Non-edible oils (e.g. jatropha and karanja oils) and waste cooking oil could be an alternative feedstock, as BD production may also provide a solution for their disposal. However, such feedstock usually possess high concentrations of free fatty acid (FFA) and moisture, hence homogeneous alkali catalyst could not be employed for their transesterification. Conventionally high FFA-containing vegetable oils (VO) are transesterified in a two-step process, involving (i) homogeneous acid (e.g., H2SO4, HCl, etc.) catalyzed pre-esterification followed by (ii) alkali (e.g., NaOH, KOH, etc.) catalyzed transesterification.4 The major drawbacks of this method are the subsequent neutralization, separation, and purification steps which are time consuming and generate huge quantity of industrial effluents. Thus a heterogeneous catalyst capable of simultaneously catalyzing esterification as well as transesterification of high FFA-containing feedstock could avoid the problems linked to the use of homogeneous catalysts.Omar and Amin5 reported a Sr/ZrO2catalyst which possesses both acidic and basic sites that is capable of simultaneously catalyzing transesterification and esterification of waste cooking oil having 2.5 wt% FFA content. The main drawback of the Sr/ZrO2 catalyst was the lower biodiesel yield (79.7%) even at high reaction temperature (115.5 °C) that using a too higher methanol to oil molar ratio (29:1). Yan et al.4 used a ZnO–La2O3 catalyst to achieve a 96% FAAE yield from waste oil with 5.4 wt% FFA at the reaction temperature of 170–200 °C. Rattanaphra et al.6 employed SO42−/ZrO2 for the transesterification of rapeseed oil having 10 wt% FFA content to achieve a 86% yield but the leaching of sulfate group into the reaction mixture remains an issue. Sreeprasanth et al.7 prepared an Fe–Zn double metal cyanide for the esterification and transesterification reaction but a decrease in biodiesel yield was observed due to the conversion of ester into FFA, caused by the hydrolytic activity of the catalyst. Kulkarni et al.8 reported tungstophosphoric acid supported on hydrous zirconia for the simultaneous esterification and transesterification of canola oil with 20 wt% FFA to achieve a 90% ester yield at high reaction temperature (200 °C). Thus most of the catalysts require high temperature and pressure for BD production from low quality feedstock having high FFA contents. Such operational conditions require complicated and costly reactors which eventually lead to the increase in BD production cost.
Another issue related to the current BD production technology is the application of methanol which is a highly toxic chemical and is itself a refinery residue.9 On the other hand, ethanol is not only non-toxic but also renewable as it is produced mainly by the fermentation of biomass. However, due to low reactivity and difficulty in the separation of FAEE from glycerol it could not be explored extensively for BD production in literature, as well as at the commercial level. SO42−/ZrO2, CaO, Zr/CaO, MgO/SBA-15, Mg2CoAl, CaO–La2O3, Ca(OCH2CH3) and calcium zincate are a few examples of literature reported heterogeneous catalysts for the ethanolysis of VOs in batch reactors.10–17 The major problems with the use of these catalysts are the requirement of high pressure and high temperature condition, and/or low conversion levels and poor reusability. To the best of our knowledge, no report is available in literature for the simultaneous esterification and transesterification of VOs with ethanol in the presence of heterogeneous catalyst.
In the present study, mixed oxides of alkaline earth metals and Zr was prepared via the co-precipitation method and employed for the ethanolysis of waste cotton seed oil. The activity of the catalysts were compared and the 2Sr:Zr-650 catalyst was found to show optimal activity in the ethanolysis. The kinetics of the ethanolysis of waste cotton seed oil was studied under optimized reaction condition. The catalyst, 2Sr:Zr-650, was able to perform the simultaneous esterification and transesterification of vegetable oil having FFA content as high as 18.1 wt%.
2. Experimental section
2.1. Materials
Zirconium oxychloride octahydrate was obtained from Sigma Aldrich while strontium nitrate, barium nitrate, calcium nitrate, magnesium nitrate, hexane, ethyl acetate, acetic acid, methanol, ethanol, ammonia (all chemicals were of analytical grade), and silica gel (TLC grade) were obtained from LobaChemie Ltd. (India) and used without further purification. n-Butylamine, trichloroacetic acid and benzene (HPLC grade) used for Hammett indicator titration were obtained from Spectrochem Pvt. Ltd. (India).Waste cottonseed oil (WO), fresh cottonseed oil (CO), karanja oil (KO) and jatropha oil (JO) used for the transesterification reactions were procured from the local shops located in Patiala and their chemical analysis is provided in Table S1 (ESI†).
2.2. Catalyst preparation
The mixed oxides strontium and zirconium with an atomic ratio of 0.3–3 (Sr/Zr) was prepared by the co-precipitation method. In a typical preparation, 5 g of ZrOCl2·8H2O and an appropriate amount of Sr(NO3)2 was mixed in 50 mL distilled water. To this 25 wt% ammonia solution was added until the pH of the mixture reached ∼10. The resulted mixture was stirred for 4 h, and filtered to obtain white precipitates which were washed with distilled water to neutralise the pH. The white solid, thus obtained, was dried at 120 °C for 24 h and finally calcined at 650 °C for 5 h. The mixed oxides of magnesium, calcium and barium with zirconium were also prepared following the same experimental procedure but by using appropriate metal precursors in place of Sr(NO)3. Prepared catalysts were labelled as xM:Zr-T, where x, M and T represents the M/Zr atomic ratio, alkaline metal and calcination temperature (°C), respectively.2.3. Characterization
Powder X-ray diffraction (XRD) patterns were recorded on a PANalytical's X'pert Pro using a monochromatic Cu Kα radiation (λ = 1.54060 Å) over a 2θ range of 5–80°. Thermo-gravimetric-differential scanning calorimetry analysis (TG-DSC) was recorded on Netzsch STA 449F3. The programmed heating ranged from 25 to 800 °C at a heating rate of 10 °C min−1 under normal atmosphere. Fourier transform-infrared spectra (FTIR) of the samples were recorded in KBr Matrix on Perkin Elmer-Spectrum 400 spectrophotometer, in the range of 400–4000 cm−1.Field emission scanning electron microscopy coupled with energy dispersive X-ray spectrometry (FESEM-EDX) was performed on JEOL JSM6510LV and transmission electron microscopy (TEM) was performed on HITACHI 7500 instruments.
The surface area of the catalysts was determined by using the adsorption desorption method at 77 K by the standard Brunauer–Emmett–Teller (BET) method using Micromeritics TriStar-3000 surface area analyzer. All samples were degassed at 473 K for 2 h under a nitrogen atmosphere to remove the physisorbed moisture from the catalysts.
Gas chromatography-mass spectroscopy (GC-MS) of FAEE was performed on Bruker GC-45X coupled with Scion MS system. Gas chromatography of FAEE samples were performed on a 15 m × 0.25 mm × 0.25 mm (RTX-5MSsil) capillary column. A sample solution of 1 μL, prepared in hexane, was injected (injection temperature 250 °C) into GC for analysis in split/splitless mode (split ratio 1:20 for 0.01 s). The flow rate of carrier gas (helium) was maintained at 1 mL min−1. The column oven temperature was increased up to 300 °C with a heating rate of 10 °C min−1. The output from the GC column entered into the ionization chamber of the mass spectrometer via a transfer line maintained at 260 °C. MS (EI 70 eV, ion source temperature 280 °C, solvent delay 2.5 min) was scanned in the m/z range of 50–800. The National Institute of Standard and Test (NIST) library match software was used to identify the FAEE. Fourier transform-nuclear magnetic resonance (FT-NMR) spectra of biodiesel and VOs were recorded on a JEOLE CS400-400 (400 MHz) spectrophotometer in CDCl3 solvent using tetramethylsilane (TMS) as an internal reference.
The metal ion concentration in FAEE and glycerol was estimated by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) on Spectro ARCOS.
The basic strengths of the catalysts (H_) were measured by the Hammett indicator benzene–carboxylic acid titration method18 using indicators namely neutral red (H_ = 6.8), bromothymol blue (H_ = 7.2), phenolphthalein (H_ = 9.3), nile blue (H_ = 10.1), tropaeolin-O (H_ = 11.1), 2,4-dinitroaniline (H_ = 15.0), and 4-nitroaniline (H_ = 18.4). The acidity of the catalysts was measured by Hammett indicator benzene–amine titration method18 using methyl red (H_ = 4.8) and methyl orange (H_ = 3.3) indicators.
2.4. Transesterification reaction and product analysis
Transesterification reactions of WO were carried out in a 50 mL two-neck round bottom flask equipped with a water-cooled reflux condenser in a water bath on a magnetic stirrer. In a typical transesterification reaction, 10 g of waste cottonseed oil was mixed with a desired molar concentration of ethanol and catalyst, and stirred at a constant speed at a desired temperature. To monitor the progress of the reaction, the samples (0.25 mL) from the reaction mixture were withdrawn every 15 min with the help of a glass dropper, centrifuged and subjected to 1HNMR and GC-MS analysis to quantify the FAEE by following the literature procedure.19,20 1H and 13C-NMR spectrum of corresponding oil and FAEE are shown in Fig. S1.†FFA conversion into FAEE was calculated21 from eqn (1).
 | (1) |
The turnover frequency (TOF) was calculated22 from eqn (2).
 | (2) |
3. Results and discussion
3.1. Catalyst characterization
 | ||
| Fig. 1 XRD pattern of (a) varying Sr:Zr atomic ratio and (b) calcination temperature (• = t-SrZrO3,■ = c-SrO,◆ = m-ZrO2,▼ = Sr(NO3)2). | ||
Comparison of the XRD patterns of 2Sr:Zr at varying calcination temperatures (350–750 °C) is shown in Fig. 1(b). At lower calcination temperatures (350–550 °C), strontium nitrate phase (Sr(NO3)2; JCPDS 87-0557) is the major phase along with m-ZrO2, t-SrZrO3 and c-SrO as minor phases. At 650 °C, due to the solid state reaction between Sr(NO3)2 and ZrO2, diffraction patterns supported the formation of t-SrZrO3 as the major phase and a further increase in calcination temperature was not found to initiate any major structural changes in catalyst structure. Thus, 650 °C was considered as the optimum temperature for catalyst preparation.
As shown in Table 1, an increase in Sr/Zr ratio was also found to increase the crystallite size of the catalyst which was probably caused by the large ionic radii of Sr (1.27 Å) instead of Zr (0.76 Å). An increase in calcination temperature (350–650 °C) was found to increase the crystallite size caused by the sintering of the catalyst particles at high calcination temperatures.
:Zr-T catalyzed transesterification
| Catalyst | Crystallite size (nm) | Basic strength | Neutral red pKBH+ = 6.8 | Bromothymol blue pKBH+ = 7.2 | Phenolphth-alein pKBH+ = 9.3 | Nile blue pKBH+ = 10.1 | Trapeolin pKBH+ = 11.1 | Total basicity (mmol g−1 of catalyst) | TOFa (h−1) |
|---|---|---|---|---|---|---|---|---|---|
| a TOF is calculated at 25% conversion level; NC = negligible conversion. Reaction conditions = ethanol to oil molar ratio of 12:1 at 75 °C reaction temperature, in the presence of 5 wt% of catalyst with respect to oil at 400 rpm stirring speed. |
|||||||||
| 0.3Sr:Zr-650 | 8.74 | 9.3 < H_<10.1 | 2.0 | 2.6 | 3.8 | 4.0 | 2.4 | 14.8 | 0.30 |
| 0.5Sr:Zr-650 | 9.88 | 9.3 < H_<10.1 | 2.2 | 2.9 | 4.1 | 4.3 | 3.2 | 16.7 | 0.35 |
| 1Sr:Zr-650 | 24.8 | 10.1 < H_<11.1 | 2.5 | 3.3 | 4.3 | 4.7 | 3.8 | 18.9 | 0.48 |
| 2Sr:Zr-650 | 27.1 | 11.1 < H_<15.0 | 3.6 | 4.3 | 5.0 | 5.5 | 4.3 | 22.7 | 0.71 |
| 3Sr:Zr-650 | 26.1 | 11.1 < H_<15.0 | 3.4 | 4.3 | 4.9 | 5.4 | 4.1 | 22.1 | 0.68 |
| 2Sr:Zr-350 | 22.8 | 7.2 < H_<9.3 | 1.2 | 1.5 | 2.3 | 2.8 | 1.8 | 9.60 | 0.28 |
| 2Sr:Zr-450 | 24.9 | 9.3 < H_<10.1 | 1.9 | 2.0 | 2.8 | 3.4 | 2.2 | 12.3 | 0.35 |
| 2Sr:Zr-550 |
26.8 | 10.1 < H_<11.1 | 2.4 | 2.7 | 3.2 | 3.7 | 3.0 | 15.0 | 0.47 |
| 2Sr:Zr-750 | 24.7 | 11.1 < H_<15.0 | 3.3 | 3.8 | 4.7 | 5.2 | 4.1 | 21.1 | 0.65 |
| 2Mg:Zr-650 |
21.8 | 9.3 < H_<10.1 | 2.2 | 3.0 | 3.6 | 2.5 | 1.8 | 13.1 | NC |
| 2Ca:Zr-650 |
22.4 | 10.1 < H_<11.1 | 2.7 | 3.6 | 3.9 | 4.2 | 3.4 | 17.8 | 0.24 |
| 2Ba:Zr-650 |
32.3 | 7.2 < H_<9.3 | 1.4 | 1.6 | 2.4 | 2.7 | 1.6 | 9.70 | NC |
:1 which is close to the theoretical value of 2:1.
 | ||
| Fig. 2 (a) SEM and (b) TEM image of 2Sr:Zr-650 catalyst. | ||
The nitrogen adsorption–desorption isotherms (Fig. S4(a), ESI†) indicate a type IV isotherm profile for 2Sr:Zr catalysts at different calcination temperatures with the hysteresis loop H3, at the relative pressure of about 0.7–1.0 which is characteristic of mesoporous materials. The non-localized density functional theory (NLDFT) and Grand Canonical Monte Carlo (GCMC) method were used to calculate the pore size distribution. Despite the smaller surface area, the catalysts have a larger pore diameter in accordance with Table S2 (ESI†). The catalyst at low calcination temperature (350 °C) shows a narrow pore size distribution with ∼9 nm pore diameter. On increasing the calcination temperature (450–550 °C), the pore diameter increases to ∼24 nm (Fig. S4(b); ESI†) and at 650 °C it reached the maximum value of ∼32 nm. At low calcination temperature, Zr(OH)4 might be occupying the pores of Sr(NO3)2/SrO to reduce the pore size of 2Sr:Zr. At high calcination temperature (650 °C), due to the solid state chemical reaction of ZrO2 with Sr(NO3)2/SrO, the pores might have been vacated to form material with relatively large pore sizes.
3.2. Structure–activity relation of catalysts
The activities of the various catalysts were compared on the basis of their total basic sites, and the TOFs of WO ethanolysis. The catalysts prepared using Mg or Ba show very little activity while the catalyst having Ca was found to be less active in comparison to the Sr/Zr catalyst. There could be two reasons behind the difference in activity, viz., (i) difference in catalyst basic strength and (ii) difference in catalyst structure. It is evident from Table 1, the basic strength of 2Sr:Zr-650 was found to be 22.7 mmol g−1 which is much higher in comparison to the 2Mg:Zr-650, 2Ca:Zr-650 and 2Ba:Zr-650 catalysts (9.7–17.8 mmol g−1).
To compare the catalyst structure, 2Mg:Zr-650, 2Ca:Zr-650, 2Ba:Zr-650 and 2Sr/Zr-650 catalysts were prepared under the same experimental conditions and their powder XRD diffraction patterns were compared. As could be seen from Fig. S5 (ESI†) in the case of Mg:Zr catalyst, diffraction patterns supported the formation of a single MgO phase, while in the case of Ba:Zr, pure BaZrO3 phase formation was observed. On the other hand, in the case of Ca:Zr and Sr:Zr catalysts, XRD diffraction patterns supported the formation of mixed phases of CaO/CaZrO3 and SrO/SrZrO3, respectively. This structural disparity might also be responsible for the variation in catalytic activity of the materials prepared with different alkaline earth metals. Nevertheless, Sr/Zr with the highest activity was studied in detail in the present manuscript. As shown in Table 1, the basic sites of the catalysts increases gradually upon the increase in the Sr/Zr ratio and calcination temperature. The optimum atomic ratio of 2 (Sr/Zr) and calcination temperature of 650 °C were able to yield the catalyst with maximum basic sites. The increase in basicity may be due to the presence of t-SrZrO3 as major phase at this ratio (Sr/Zr = 2), which is reported to be an active species for transesterification reaction.25 A further increase in strontium to zirconium ratio (≥3) was not found to increase the activity of the catalyst to a significant extent as the total basic site remains almost similar to the catalyst having a Sr/Zr ratio of 2.
Similarly, by increasing the calcination temperature, the basic site as well as activity of the catalyst was found to increase and a maximum activity was observed at 650 °C. At low calcination temperature (350-550 °C), the lesser activity is due to the presence of strontium nitrate which eventually decomposed into SrO at 650 °C. The reaction between SrO and ZrO2 leads to the formation of SrZrO3 which causes the enhancement in catalytic activity. Since 2Sr:Zr-650 catalyst was found to catalyze the reaction rate to the maximum extent, it was selected for optimizing the other reaction parameters for the transesterification reaction.
Usually catalysts with both acidic as well as basic sites were found to show simultaneous esterification and transesterificationactivity.4 The catalyst, 2Sr:Zr-650, was also found to have moderate acidic sites (3.9 mmol g−1 of catalyst; acidic strength 3.3 < H_ < 4.8) which can catalyze fatty acid esterification. In order to show the esterification activity of 2Sr:Zr-650 catalyst, esterification of oleic acid with ethanol (1:12 molar ratio) was performed at 75 °C. As could be seen from Fig. S6 (ESI†), in 3.5 h of reaction duration a 70.6% ethyl oleate yield was obtained which proves the esterification activity of the catalyst. Moreover, biodiesel prepared from high FFA-containing oil (up to 18 wt%) was found to show significantly lesser acid value (0.4 mg KOH/g), which further supports the esterification of FFA present in triglyceride. To the best of our knowledge this is the first report for the simultaneous esterification and transesterification of high FFA-containing triglycerides with ethanol in the presence of heterogeneous catalyst.
3.3. Optimization of the reaction parameters
In order to optimize the reaction parameters to achieve better catalytic performance, the following reaction parameters were studied (Fig. S7, ESI†): (i) stirring speed (100–700 rpm), (ii) catalyst concentration (1–6 wt%), (iii) ethanol/oil molar ratio (3–15), and (iv) reaction temperature (35–85 °C). On the basis of this study, a 12:1 ethanol to oil molar ratio at 75 °C in the presence of 5 wt% catalyst (with respect to oil) at a stirring speed of 400 rpm were found to be the optimum conditions for the 2Sr:Zr-650 catalyzed transesterification of WO.
A comparison between the ethanolysis activity of 2Sr:Zr-650 catalyst with literature reports is given in Table 2. As could be seen from the comparison, the use of present catalyst for VO ethanolysis is beneficial as it not only requires lesser alcohol to oil molar ratio, lower reaction temperature but also demonstrates moderate stability and reusability.
| Catalyst | Oil | C.A (wt%) | R.T (°C) | E:O |
Reaction time (h) | FAAE yield (%) | Reusability (FAEE yield after last run) | Ref. |
|---|---|---|---|---|---|---|---|---|
| C.A – catalyst amount; R.T – reaction temperature; E:O – ethanol to oil molar ratio; N.R – not reported; a -under high pressure in parr batch reactor. |
||||||||
| SO42−/ZrO2a | Soybean oil | 5 | 120 | 20:1 |
1 | 92 | 4 (15) | 10 |
| CaO | Sunflower oil | 20 | 75 | 18:1 |
6 | 100 | N.R | 11 |
| Zr/CaO | Jatropha oil | 5 | 75 | 21:1 |
7 | >99 | N.R | 12 |
| MgO/SBA-15a | Edible oil | 2 | 220 | 6:1 |
5 | 96 | N.R | 13 |
| Mg2CoAla | Rapessed oil | 2 | 200 | 16:1 |
18 | 97 | 7 (97) | 14 |
| CaO–La2O3 | Soybean oil | 8 | 65 | 10:1 |
6 | 71.6 | N.R | 15 |
| Ca(OCH2CH3) | Soybean oil | 3 | 75 | 12:1 |
3 | 91.8 | N.R | 16 |
| Calcium zincate | Sunflower oil | 6 | 78 | 20:1 |
3 | ≈90 | 3 (40) | 17 |
| 2Sr:Zr | Waste cotton seed oil | 5 | 75 | 12:1 |
7 | >99 | 4 (98) | Present study |
3.4. Effect of moisture and FFA
Homogeneous catalysts require costlier refined feedstock for the transesterification reactions as the presence of >0.3 wt% moisture and/or >0.5 wt% FFA contents in feedstock leads to the formation of soap instead of biodiesel.4 In the present study, the WO employed as feedstock was found to have 4.7 and 0.27 wt% FFA and moisture contents, respectively. When the transesterification of the same oil in the presence of homogeneous base catalyst (NaOH) was performed, catalyst deactivation occurred due to soap formation. However, >99% FAEE yield was obtained in the 2Sr:Zr-650 catalyzed transesterification of WO. In order to determine the maximum moisture resistance of the catalyst, the transesterification reactions of WO were performed by adding up to 4.5 wt% (water/oil) of water in reaction mixture. The 2Sr:Zr-650 catalyst was found to be effective for thetransesterification of WO having up to 2.0 wt% of moisture contents as shown in Fig. 3(a). However, a further increase in water concentration (2.0–4.5 wt%) was found to decrease the catalyst efficiency. This could be attributed to the change of Lewis basic sites (–O–) into Brønsted basic sites (–OH) due to the reaction of water with catalyst. The Brønsted basic sites were found to be less active towards the transesterification than corresponding Lewis basic sites.4,12 The conversion of oxides into respective hydroxides is also evident from the comparison of XRD of neat and moisture exposed catalysts as shown in Fig. 3(b).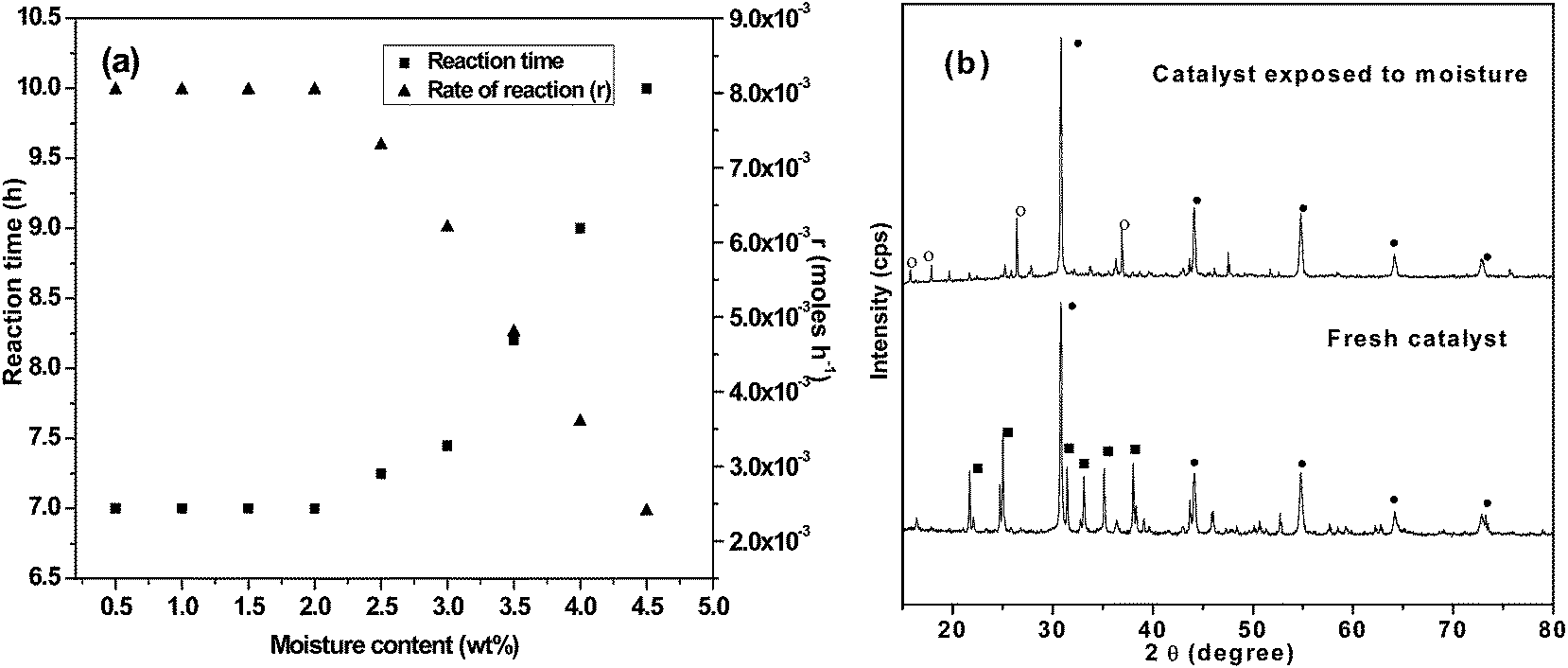 | ||
| Fig. 3 (a) Effect of moisture content on the 2Sr:Zr-650 catalyzed transesterification of WO (reaction time is the time required to achieve >99% FAEE yield); (b) comparison of XRD spectra of fresh catalyst and catalyst exposed to water (• = SrZrO3, ○ = Sr(OH)2). | ||
Non-edible or waste cooking oils are usually found to have FFA contents in relatively higher amount (4.6–18.1 wt% in the present study). Conventionally, the transesterification of high FFA-containing vegetable oils is performed in a two-step process involving acid-catalyzed pre-esterification step in the presence of an acid catalyst followed by transesterification in the presence of alkali catalyst.4 The 2Sr:Zr-650 catalyst was found to show esterification as well as transesterification activity as discussed in the previous sections. In order to demonstrate the simultaneous esterification as well as transesterification activity of the catalyst in one-pot, transesterification reactions of CO, WO, JO and KO (having 1–18.1 wt% FFA) were performed with ethanol as shown in Fig. 4. Although the catalyst was found to be effective for the transesterification of high FFA-containing VOs, a decrease in TOF was observed with the increase in FFA contents. This may be due to the strong interactions of highly polar acetate (–COO−) functional group with catalyst (Sr2+/Zr4+) to result in the partial blocking of the active sites.
 | ||
| Fig. 4 Effect of FFA contents on the 2Sr:Zr-650 catalyzed transesterification of different feedstock (reaction time is the time required to achieve >99% FAEE yield). Reaction conditions: ethanol to oil molar ratio of 12:1 at 75 °C reaction temperature in presence of 5 wt% (catalyst/oil) catalyst. | ||
3.5. Reusability
Commercial success of BD production technologies depends on its production cost. Catalyst reusability could reduce the BD production cost to a significant extent. In order to study the stability and reusability of catalyst, it was separated from the reaction mixture by centrifugation, washed with hexane and finally regenerated at the calcination temperature of 650 °C. The regenerated catalyst was employed in five successive catalytic cycles under the same experimental and regeneration methods. As could be seen from Fig. 5, no significant loss in catalytic activity was observed during first four runs; however, a partial decrease in activity was observed in the fifth cycle. | ||
| Fig. 5 Reusability study of catalyst. | ||
The structural changes and/or leaching of the active catalyst ingredient or blockage/poisoning of active sites could be the reasons behind the loss of activity in heterogeneous catalysts.26 To identify the structural changes, XRD patterns of fresh and regenerated 2Sr:Zr-650 catalysts are compared in Fig. 6(a). In the XRD pattern of regenerated 2Sr:Zr-650 catalyst, diffraction peaks due to SrO were no longer found. The crystallite size of regenerated catalyst was found to be bigger (30.2 nm) than fresh catalyst (27.1 nm) to support partial agglomeration of catalyst during re-calcination process.
 | ||
| Fig. 6 Comparison of (a) XRD and (b) FT-IR of fresh and used catalyst. (■ = SrO; • = SrZrO3). | ||
As shown from Fig. 6(b), in FTIR spectrum of fresh catalyst, the stretching vibrations associated with Sr–O and Zr–O bonds appear at 550 and 648 cm−1, respectively.25 The bands appeared at 1453 and 1638 cm−1 supported the presence of hydroxide and water molecule due to catalyst surface hydration. In regenerated catalyst, new bands appeared at 909 and 974 cm−1, in addition to the original bands, to support the changes in catalyst structure which is also consistent with the XRD analysis of the reused catalyst. The spectrum of the regenerated catalyst did not show vibrations corresponding to any adsorbed organic molecules to indicate that FAEE or glycerol was not accumulated on the surface of regenerated catalyst.
The metal analysis supported the presence of metal in FAEE (Sr = 1.6 ppm and Zr = 0.8 ppm) as well as in glycerol (Sr = 4.6 ppm and Zr = 3.9 ppm). Thus metal is gradually lost during the catalytic run, which could be another reason for the loss of activity. To ensure the heterogeneous nature of the catalyst, and to prove that leached metal ion has not acted like a homogeneous catalyst, a hot filtration test was performed under optimized reaction conditions. After 1.5 h of reaction, the catalyst was removed by filtration and reactants were heated again for an additional 3.5 h. As could be seen from Fig. 7, no significant gain in FAEE yield was obtained after the catalyst removal to prove (i) a truly heterogeneous mode of action of Sr:Zr catalyst and (ii) that leached metal has no contribution in catalytic activity.
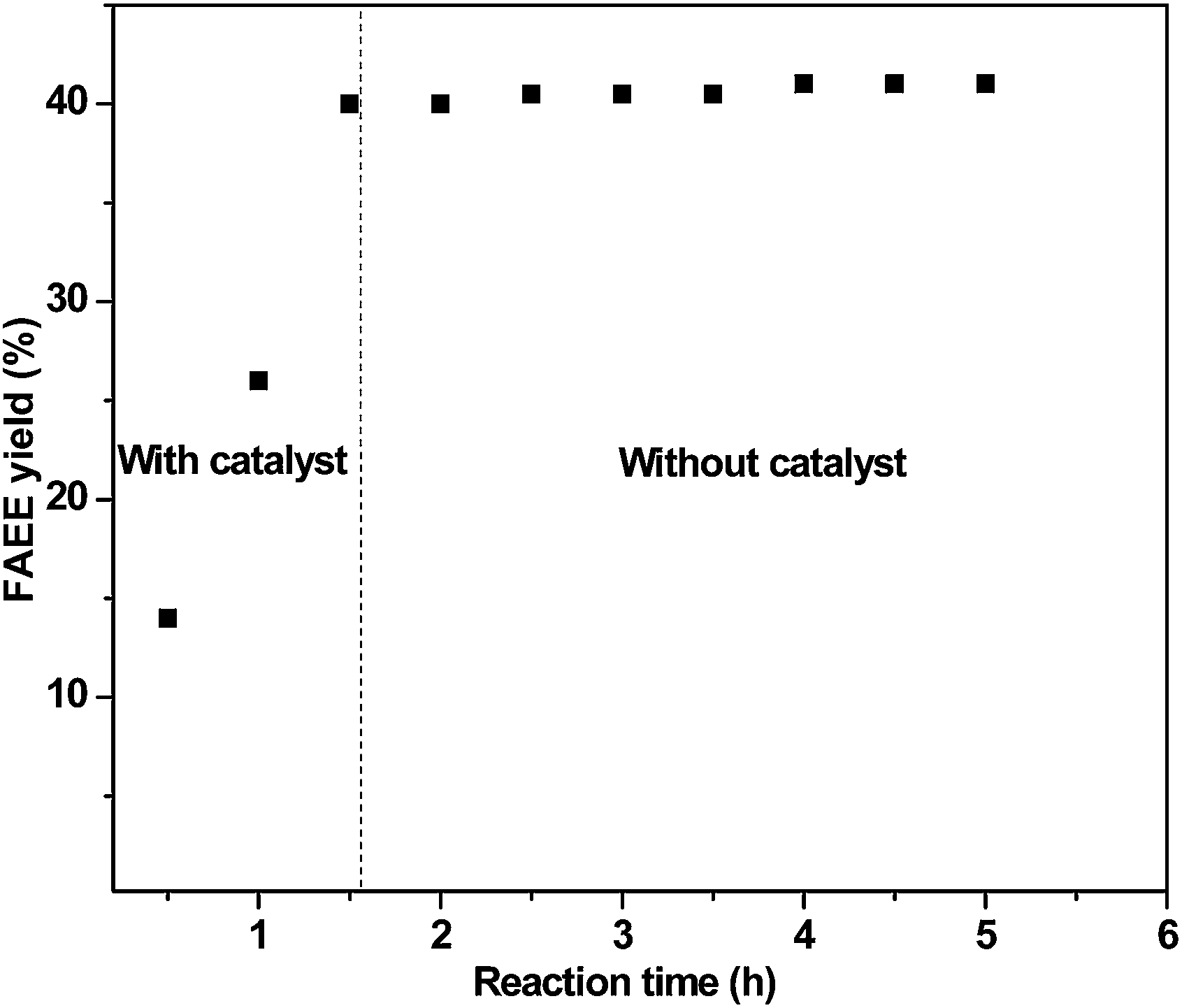 | ||
| Fig. 7 Hot filtration test for 2Sr:Zr-650 catalyzed transesterification. | ||
Therefore, the gradual loss of the catalytic activity could be attributed to (i) structural changes in catalyst structure and (ii) partial loss of the Sr and Zr from 2Sr:Zr-650 upon repeated use.
3.6. Koros–Nowak criterion test
The absence of internal transfer limitations was demonstrated by the Koros–Nowak criterion test.27,28 In the present study the reactions were carried out in the presence of two catalysts viz., 5 wt% of 2Sr:Zr-650 or 6 wt% of 1Sr:Zr-650 catalyst dosages to maintain the same fractional exposures of active (basic) sites. At similar conversion levels the TOF of both catalysts were found to be almost similar as shown in Fig. S8 (ESI†). Thus the Sr:Zr catalyzed reaction is free from mass diffusion limitations and is a chemically controlled reaction.3.7. Kinetics and thermodynamic study
Transesterification is generally assumed to follow pseudo-first-order kinetics as alcohol in such reactions are employed in excess to the required stoichiometric amount of 3:1 alcohol to oil molar ratio.27 The present work also optimized alcohol to VO molar ratio at 12:1 and hence, the kinetics of 2Sr:Zr-650 catalyzed ethanolysis were studied by following (pseudo) first order eqn (3):| −ln(1 − X) = kt | (3) |
The linear nature of the plot between −ln(1 − X) vs. t plot supported that the reaction has followed the (pseudo) first order rate law (Fig. 8(a)). The apparent first order rate constant form the plot was found to be 0.648 h−1 at 75 °C.
 | ||
| Fig. 8 Kinetic study of transesterification of WO with ethanol over 2Sr:Zr-650 catalyst. (a) Plots of −ln(1 − X) vs. time at different temperatures; (b) Arrhenius plot of lnk vs. 1/T. Reaction conditions: ethanol to oil molar ratio of 12:1 and 5 wt% of 2Sr:Zr-650with respect to oil. | ||
To calculate the activation energy, reactions were carried out in temperatures range of 35–75 °C. The Arrhenius equation was employed to estimate the activation energy (Ea) and pre-exponential factor (A) following29 eqn (4):
|
lnk = lnA − Ea/RT
| (4) |
A plot between lnk vs. 1/T is shown in Fig. 8(b), and the values of Ea and A from the plot was found to be 48.17 kJ mol−1 and 6.83 × 106 h−1. The observed Ea value in the present study (48.17 kJ mol−1) was found within the range of the reported values (26–82 kJ mol−1) for the transesterification reaction catalyzed by heterogeneous catalysts.12
A value of Ea > 25 kJ mol−1 also supported the observation that the 2Sr:Zr-650 catalyzed transesterification is a chemically controlled reaction and not controlled by mass transfer limitations.30
Thermodynamic analysis was addressed for evaluating the enthalpy (ΔH‡), entropy (ΔS‡), and the Gibb's free energy of activation (ΔG‡), which are important features for interpreting the behaviour of transesterification reactions. In this regard, activation complex theory, developed by Eyring, was applied to evaluate thermodynamic parameters from temperature-dependent rate constants. These parameters are calculated31 from the Eyring–Polanyi equation (eqn (5)) which is analogous to the Arrhenius equation:
 | (5) |
Taking the natural logarithm of eqn (5) and substituting the value of ΔG‡ = ΔH‡ − TΔS‡, eqn (6) is obtained:
 | (6) |
Fig. 9 depicts the Eyring plot of transesterification and the value of ΔH‡ was found to be 45.97 kJ mol−1 indicating that energy input (heat) from external source is required to raise the energy level and transform the reactants to their transition state. The value of ΔS‡ was found to be −121.37 J mol−1 K−1 and this negative value suggests an associative mechanism in which reactant species have joined together to form a more ordered transition state. The ΔG‡ value was found to be 88.23 kJ mol−1 to indicate the unspontaneous nature of the reaction having higher energy level of transitionstate than reactant species.
 | ||
| Fig. 9 The Eyring plot of 2Sr:Zr-650 catalyzed transesterification of WO. | ||
3.8. GC/MS analysis of FAEE
Physicochemical properties such as viscosity, freezing point, and cold flow properties of BD depend upon its chemical composition. GC/MS analysis was performed to quantify the FAEE yield as well as FAEE chemical composition. Seven peaks were observed in GC chromatogram (Fig. S9†) of FAEE prepared from WO. Each peak corresponds to a FAEE which was identified on the basis of MS pattern from the library match software (NIST) and the component corresponding to individual peak is listed in Table 3. The total FAEE contents obtained through area normalization method was found to be 99.61 wt%, out of which total saturated and unsaturated fatty acid alkyl ester content were found to be 31.32 and 68.29 wt%, respectively. A higher unsaturated fatty acid alkyl ester content reflects a better cold flow property of BD even at low temperatures.32| S. no. | Retention time (min) | Composition; molecular formula | Corresponding acid (Cx:y) |
Wt% |
|---|---|---|---|---|
| a Cx:y = x is number of carbon atom, y is number of double bonds. |
||||
| 1 | 7.66 | Myristic acid ethyl ester; C16H32O2 | Myristic acid (C14:0) |
0.86 |
| 2 | 8.57 | Palmitoleic acid ethyl ester; C18H34O2 | Palmitoleic acid (C16:1) |
0.62 |
| 3 | 8.68 | Palmitic acid ethyl ester; C18H36O2 | Palmitic acid (C16:0) |
26.64 |
| 4 | 9.46 | Linoleic acid ethyl ester; C20H36O2 | Linoleic acid (C18:2) |
37.77 |
| 5 | 9.49 | Oleic acid ethyl ester; C20H38O2 | Oleic acid (C18:1) |
29.90 |
| 6 | 9.61 | Stearic acid ethyl ester; C20H40O2 | Stearic acid (C18:0) |
3.44 |
| 7 | 9.90 | Eicosanoic acid ethyl ester; C22H44O2 | Eicosanoic acid (C20:0) |
0.38 |
3.9. Physicochemical properties of FAEE
The fatty acid alkyl esters must satisfy ASTM D6751 or EN 14214 specifications before they could be employed for commercial applications as a fuel. In order to study the physicochemical properties, FAEE were prepared by the transesterification of WO with ethanol under optimized reaction conditions. After the completion of the reaction, the catalyst was separated through filtration and the liquid phase thus obtained was kept in a separating funnel for 24 h to separate the lower glycerol layer from upper FAEE layer. Excess alcohol from ester layer was recovered with the help of a rotary evaporator. A number of physicochemical properties of the prepared FAEE were studied by following the standard test methods and were compared with EN 14214 specifications. As could be seen from Table 4, the observed values of the studied properties were found within the acceptable limits of European standards. Specifically, the total metal concentration in crude FAEE was found to be less than the acceptable limit and hence, biodiesel washing with water would not be required which could avoid the generation of huge amount of industrial effluents which is inevitable in the case of homogeneous catalytic process.| S.No. | Parameters | Units | FAEE | EN14214 | Test method |
|---|---|---|---|---|---|
| 1 | Ester content | % | >99% | ≥96.5 | 1H-NMR, GC-MS |
| 2 | Flash point | °C | 120 | 100–170 | ASTM D93 |
| 3 | Pour point | °C | 3 | −5 to10 | ASTM D2500 |
| 4 | Water content | % | 0.25 | ≤0.5 | ASTM D2709 |
| 5 | Kinematic viscosity at 40 °C | cSt | 4.63 | 1.9–6.0 | ASTM D445 |
| 6 | Density at 31 °C | kg mm−3 | 875 | 860–900 | ISI448 P:32 |
| 7 | Ash | % | 0.01 | 0.02 | ASTM D874 |
| 8 | Iodine value | mg of I2/g of sample | 88.4 | ≤120 | 1H-NMR33 |
| 9 | Acid value | mg of KOH/g of sample | 0.4 | 0.8 | ASTM D664 |
| 10 | Saponification value | mg of KOH/g of sample | 181.23 | — | ASTM D5558 |
| 11 | Sr/Zr | ppm | 1.6/0.8 | ≤5 (total metal) | ICP-AES |
4. Conclusions
In the present work, 2Sr:Zr-650 catalyst has been prepared as ∼25–30 nm sized particles as revealed by powder XRD and TEM techniques. The catalyst was found to be effective for the one-pot esterification and transesterification of a variety of feedstock having up to 2.0 and 18 wt% of moisture and FFA contents, respectively. Under optimized reaction conditions (catalyst concentration 5 wt%, ethanol to oil molar ratio of 12:1 at 75 °C and 400 rpm) the transesterification of waste cottonseed oil was found to follow (pseudo) first order rate law. Activation and Gibbs free energy for the ethanolysis of waste cottonseed oil was found to be 48.17 and 88.23 kJ mol−1, respectively. The catalyst was recovered and recycled without any significant loss in activity during first four catalytic cycles. The leaching test supports the negligible homogeneous contribution in catalytic activity, and KN test demonstrated that the activity is free from transport phenomenon. A number of physicochemical properties of the FAEE prepared from waste cottonseed oil were studied, and observed values were found within the limits of EN 14214 specifications.
Acknowledgements
We acknowledge CSIR (01(2503)/11/EMR-II) and DRDO (ERIP/ER/1103933/M/01/1453) for financial support. We are also thankful to the School of Chemistry and Biochemistry (Thapar University, Patiala) and DST-FIST for GC-MS, SAIF (Panjab University, Chandigarh) for powder XRD and FT-IR, IIT Ropar for SEM, SAI laboratories (Thapar University, Patiala) for NMR, and Avansa Technology and Services (Kanpur) for surface area analysis. We are thankful to Dr Nitin Kumar Singhal (NABI, Mohali) for ICP-AES and Dr B. N.Chudasama for TGA-DSC study.References
- Q. Liu, B. Wang, C. Wang, Z. Tian, W. Qu, H. Ma and R. Xu, Green Chem., 2014, 16, 2604 RSC
.
- A. P. S. Dias, J. Bernardo, P. Felizardo and M. J. N. Correia, Fuel Process. Technol., 2012, 102, 146 CrossRef CAS PubMed
- N. Kaur and A. Ali, Energy Sources, Part A, 2013, 35, 184 CrossRef CAS
- S. Yan, S. O. Salley and K. Y. Simon, Appl. Catal., A, 2009, 353, 203 CrossRef CAS PubMed
- W. N. N. W. Omar and N. A. S. Amin, Fuel Process. Technol., 2011, 92, 2397 CrossRef PubMed
- D. Rattanaphra, A. Harvey and P. Srinophakun, Top. Catal., 2010, 53, 773 CrossRef CAS
- P. S. Sreeprasanth, R. Srivastava, D. Srinivas and P. Ratnasamy, Appl. Catal., A, 2006, 314, 148 CrossRef CAS PubMed
- M. G. Kulkarni, R. Gopinath, L. C. Meher and A. K. Dalai, Green Chem., 2006, 8, 1056 RSC
- C. Brunschwig, W. Moussavou and J. Blin, Prog. Energy Combust. Sci., 2012, 38, 283 CrossRef CAS PubMed
- C. M. Garcia, S. Teixeira, L. L. Marciniuk and U. Schuchardt, Bioresour. Technol., 2008, 99, 6608 CrossRef CAS PubMed
- O. S. Stamenkovic, A. V. Velickovic and V. B. Veljkovic, Fuel, 2011, 90, 3141 CrossRef CAS PubMed
- N. Kaur and A. Ali, Fuel Process. Technol., 2014, 119, 173 CrossRef CAS PubMed
- E. Li and V. Rudolph, Energy Fuel, 2008, 22, 145 CrossRef CAS
- E. Li, Z. P. Xu and V. Rudolph, Appl. Catal., B, 2009, 88, 42 CrossRef CAS PubMed
- M. Kim, C. D. Maggio, S. Yan, S. O. Salley and K. Y. S. Ng, Appl. Catal., A, 2010, 378, 134 CrossRef CAS PubMed
- X. Liu, X. Piao, Y. Wang and S. Zhu, Energy Fuel, 2008, 22, 1313 CrossRef CAS
- J. M. R. Caballero, J. S. Gonzalez, J. M. Robles, R. M. Tost, M. L. A. Castillo, E. V. Alonso, A. J. Lopez and P. M. Torres, Fuel, 2013, 105, 518 CrossRef PubMed
- T. Yamanaka and K. Tanabe, J. Phys. Chem., 1975, 79, 2409 CrossRef CAS
- G. F. Ghesti, J. L. Macedo, I. S. Resck, J. A. Dias and S. C. L. Dias, Energy Fuel, 2007, 21, 2475 CrossRef CAS
- Y. Li, X. D. Zhang, L. Sun, M. Xu, W. G. Zhou and X. H. Liang, Appl. Energy, 2010, 87, 2369 CrossRef CAS PubMed
- J. M. Marchetti and A. F. Errazu, Biomass Bioenergy, 2008, 32, 892 CrossRef CAS PubMed
- G. Tao, Z. Hua, Z. Gao, Y. Chen, L. Wang, Q. He, H. Chena and J. Shi, RSC Adv., 2012, 2, 12337 RSC
- S. Sankaranarayanan, C. A. Antonyraj and S. Kannan, Bioresour. Technol., 2012, 109, 57 CrossRef CAS PubMed
- P. D. Patil and S. Deng, Energy Fuel, 2009, 23, 4619 CrossRef CAS
- J. R. O. Lima, Y. A. Ghani, R. B. Silva, F. M. C. Batista, R. A. Bini, L. C. Varanda and J. E. Oliveira, Appl. Catal., A, 2012, 445–446, 76 CrossRef CAS PubMed
- D. Singh, R. Bhoi, A. Ganesh and S. Mahajani, Energy Fuels, 2014, 28, 2743 CrossRef CAS
- R. Song, D. Tong, J. Tang and C. Hu, Energy Fuels, 2011, 25, 2679 CrossRef CAS
- R. J. Madon and M. Boudart, Ind. Eng. Chem. Fundam., 1902, 21, 438 CrossRef
- J. M. Balbino, E. W. D. Menezes, E. V. Benvenutti, R. Cataluna, G. Ebelinga and J. Dupont, Green Chem., 2011, 13, 3111 RSC
- A. Patel and V. Brahmkhatri, Fuel Process. Technol., 2013, 113, 141 CrossRef CAS PubMed
- L. K. Ong, A. Kurniawan, A. C. Suwandi, C. X. Lin, X. S. Zhao and S. Ismadji, J. Supercrit. Fluids, 2013, 75, 11 CrossRef CAS PubMed
- E. Lotero, Y. Liu, D. E. Lopez, K. Suwannakarn, D. A. Bruce and J. G. Goodwin, Ind. Eng. Chem. Res., 2005, 44, 5353 CrossRef CAS
- R. Kumar, V. Bansal, M. B. Patel and A. S. Sarpal, Energy Fuels, 2012, 26, 7005 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07178f |
| This journal is © The Royal Society of Chemistry 2014 |