
Synthesis and characterization of amphiphilic triblock Copolymers with Identical compositions but different block sequences†
Abstract
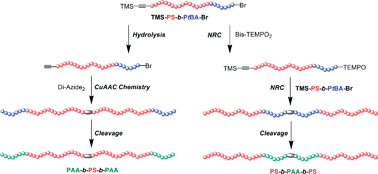
The amphiphilic triblock copolymers poly(acrylic acid)-b-poly(styrene)-b-poly(acrylic acid) (PAA-b-PS-b-PAA) and PS-b-PAA-b-PS with identical compositions but different block sequences were synthesized by a combination of an atom transfer radical polymerization (ATRP) mechanism and a nitroxide radical coupling (NRC) reaction or copper-catalyzed azide/alkyne click (CuAAC) chemistry. Firstly, the diblock copolymers TMS–![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) –PS-b-PtBA-Br were prepared by sequential ATRP of styrene (St) and tert-butyl acrylate (tBA) monomers from trimethylsilyl propargyl 2-bromoisobutyrate (TMS-PgBiB) initiator. Then, the triblock copolymers PS-b-PtBA-b-PS were prepared by NRC reaction between TMS––PS-b-PtBA-Br and coupling agent bis[4-(2,2,6,6-tetramethylpiperidine-1-oxyl)] succinate (Bis-TEMPO2). And the triblock copolymers PtBA-b-PS-b-PtBA were obtained by CuAAC chemistry between Alkynyl-PS-b-PtBA-Br and a coupling agent 1,4-diazidobutane (Di-Azide2). The target triblock copolymers PAA-b-PS-b-PAA and PS-b-PAA-b-PS were finally derived from the cleavage of the corresponding PS-b-PtBA-b-PS and PtBA-b-PS-b-PtBA. The self-assembly behaviour was preliminarily studied by FLS, FESEM and DLS instruments, and the results showed that the PS-b-PAA-b-PS and PAA-b-PS-b-PAA could give distinct critical micelle concentration (cmc) values and different sizes of micelles in water.
–PS-b-PtBA-Br were prepared by sequential ATRP of styrene (St) and tert-butyl acrylate (tBA) monomers from trimethylsilyl propargyl 2-bromoisobutyrate (TMS-PgBiB) initiator. Then, the triblock copolymers PS-b-PtBA-b-PS were prepared by NRC reaction between TMS––PS-b-PtBA-Br and coupling agent bis[4-(2,2,6,6-tetramethylpiperidine-1-oxyl)] succinate (Bis-TEMPO2). And the triblock copolymers PtBA-b-PS-b-PtBA were obtained by CuAAC chemistry between Alkynyl-PS-b-PtBA-Br and a coupling agent 1,4-diazidobutane (Di-Azide2). The target triblock copolymers PAA-b-PS-b-PAA and PS-b-PAA-b-PS were finally derived from the cleavage of the corresponding PS-b-PtBA-b-PS and PtBA-b-PS-b-PtBA. The self-assembly behaviour was preliminarily studied by FLS, FESEM and DLS instruments, and the results showed that the PS-b-PAA-b-PS and PAA-b-PS-b-PAA could give distinct critical micelle concentration (cmc) values and different sizes of micelles in water.
 Please wait while we load your content...
Please wait while we load your content...

