
Upgrading malic acid to bio-based benzoates via a Diels–Alder-initiated sequence with the methyl coumalate platform†
Abstract
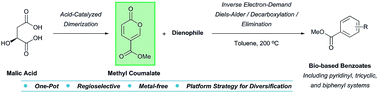
The conversion of naturally-occurring malic acid to the 2-pyrone methyl coumalate was optimized using a variety of acid catalysts. Coupling methyl coumalate with electron-rich dienophiles in an inverse electron-demand Diels–Alder (IEDDA)/decarboxylation/elimination domino sequence resulted in an investigation of the scope and limitations of the methodology. The thermal, metal-free, and one-pot procedure allows regioselective access to diverse aromatic compounds including tricyclic, biphenyl, and pyridinyl systems for elaboration. A comparison with analogous pyrones demonstrates the striking efficacy of methyl coumalate as a versatile platform for the generation of biorenewable functionalized benzoates.
 Please wait while we load your content...
Please wait while we load your content...

