
Effect of ceria morphology on the activity of MnOx/CeO2 catalysts for the catalytic combustion of chlorobenzene
Abstract
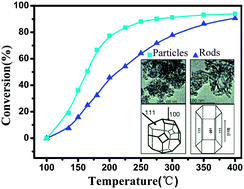
The effect of CeO2 morphology on the performance of a MnOx/CeO2 catalyst was investigated for the catalytic combustion of chlorobenzene (CB), which was used as a model compound for chlorinated volatile organic compounds (CVOCs). The catalytic activity tests revealed that MnOx/CeO2 nanoparticles (NPS) achieved relatively higher CB conversions than MnOx/CeO2 nanorods (NR). The MnOx/CeO2 catalysts were characterized by X-ray diffraction (XRD), Raman spectroscopy, Brunauer–Emmett–Teller (BET) N2 adsorption, transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS) and hydrogen temperature-programmed reduction (H2-TPR). The characterization of the MnOx/CeO2 catalysts indicated that CeO2-NPS had a higher exposure of the (100) crystal plane and possessed more Mn4+ species, oxygen vacancies and surface-adsorbed oxygen. It was suggested that the CeO2-NPS had a stronger interaction with MnOx species, which resulted in greater catalytic activity in the combustion of CB. The catalytic activity of MnOx/CeO2-NPS could be attributed to higher concentrations of Mn4+ species, oxygen vacancies, and surface-adsorbed oxygen, which were associated with the exposed (100) crystal planes. Therefore, these results demonstrated that the catalytic performance of the MnOx/CeO2 catalyst was greatly affected by the CeO2 morphology. Therefore, catalytic materials that have increased activity can be obtained through morphology-controlled synthesis.
 Please wait while we load your content...
Please wait while we load your content...

