
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient synthesis of pyrene-1-carbothioamides and carboxamides. Tunable solid-state fluorescence of pyrene-1-carboxamides†
Anna
Wrona-Piotrowicz
a,
Janusz
Zakrzewski
*a,
Rémi
Métivier
*b,
Arnaud
Brosseau
b,
Anna
Makal
c and
Krzysztof
Woźniak
c
aDepartment of Organic Chemistry, Faculty of Chemistry, University of Łódź, Tamka 12, 91-403 Łódź, Poland. E-mail: janzak@uni.lodz.pl
bPPSM, ENS Cachan, CNRS, UniverSud, 61 av President Wilson, 94230 Cachan, France. E-mail: metivier@ppsm.ens-cachan.fr
cFaculty of Chemistry, Warsaw University, Pasteura 1, 02-093 Warszawa, Poland
First published on 23rd October 2014
Abstract
Pyrene reacts with potassium thiocyanate and organic isothiocyanates in the presence of trifluoromethanesulfonic acid to afford primary and secondary pyrene-1-carbothioamides in high yields. These compounds were efficiently oxidatively desulfurized with Oxone® to the corresponding carboxamides. The amides display solid-state fluorescence with quantum efficiencies up to 62%, originating from monomers, aggregates (such as preformed dimers), and/or excimers, depending on the substituent at the nitrogen atom. Single crystal X-ray diffraction characterization of one highly emissive compound supports this assumption.
Introduction
Pyrene derivatives have attracted significant interest as materials for organic electronics,1 fluorescent molecular probes,2–4 monomers,5 triplet sensitizers6 and sensors.3,7–9 Considerable attention has been paid to the development of synthetic routes to pyrenes bearing various substituents1,10,11 and to studies on their fluorescence properties.11–13Several recent works have focused on environmentally-responsive emission of pyrene-1-carboxamides which is strongly dependent on the substituents at the nitrogen atom.12,14–16 Moreover, these compounds were used in the synthesis of polymers with tunable luminescent properties17 and in the labelling of nucleosides.7,8,18–22
In contrast to amides, pyrene-1-carbothioamides are only weakly fluorescent23,24 because of the internal quenching effect of the thiocarbonyl group. Since some chemical species desulfurize thioamides (e.g. Oxone®, H2O2 in the presence of Zr4+) to fluorescent amides, pyrene thioamides were used as fluorescent “turn-on” sensors of such species23,24.
The reported synthetic route to pyrene-1-carboxamides relies on the reaction of pyrene-1-carboxylic acid chloride with amines. The amides can then be transformed into thioamides via a reaction with Lawesson reagent.17,23,24
In this paper we report an efficient method of synthesis of primary and secondary pyrene-1-carbothioamides directly from pyrene via a Friedel–Crafts-type reaction with potassium thiocyanate and organic isothiocyanates, respectively. The thioamides were then desulfurized into the corresponding amides using Oxone®. Furthermore, we discovered that most of the synthesized amides are fluorescent not only in solution, as was reported earlier, but also in the solid state. Solid-state emission is a phenomenon of great practical importance,25–27 but is relatively rare in the case of planar aromatic compounds such as pyrene and its derivatives. We found that the solid-state emissive properties of synthesized pyrene-1-carboxamides strongly depend on the substituent on the nitrogen atom and for bulky substituents fluorescence quantum yields as high as 60% were obtained.
Results and discussion
A Friedel–Crafts-type reaction of organic isothiocyanates with aromatic compounds is a straightforward synthetic route to secondary thioamides.28–31 We recently reported that a reaction of pyrene with ethoxycarbonylisothiocyanate afforded N-ethoxycarbonyl-pyrene-1-carbothioamide in 95% yield.32 Primary thioamides were obtained in a reaction of electron-rich arenes (1,2-diethoxybenzene, ferrocene) with potassium thiocyanate in methanesulfonic acid.28,33,34Herein we report that pyrene reacts with potassium thiocyanate in the presence of 4 equiv. of TfOH to afford the primary thioamide 1a in 63% yield (Scheme 1). The reaction was performed in dichloromethane at room temperature. Under the same conditions, reaction of pyrene with alkyl- and aryl isothiocyanates afforded secondary thioamides 1b–j in 78–96% yield. Thioamides 1a–j were conveniently isolated, after quenching the reaction mixtures with water and extraction, via column chromatography on silica gel.
 | ||
| Scheme 1 Synthesis of thioamides 1a–j and amides 2a–j. | ||
After having elaborated an efficient direct method for thioamidation of pyrene, we explored the possibility of extending this method to the synthesis of pyrenyl amides using isocyanates instead of isothiocyanates. We chose the reaction of pyrene with phenyl isocyanate as a model. Unfortunately, we found that this reaction provided the expected N-phenylpyrene-1-carboxamide only in low (∼30%) yield. Because of this unsatisfactory result, we changed our approach in order to find another synthetic route to pyrenyl amides.
Since a variety of simple and efficient methods of desulfurization of thioamides to amides has been reported in the literature,35 we decided to check whether this transformation could be applied to the synthesis of pyrenyl amides.
We decided to use a commercially available and inexpensive oxidant, Oxone® (2KHSO5·KHSO4·K2SO4), for the desulfurization of thioamides 1a–j. This reagent has already been used for desulfurization of other thioamides.14,15,36,37 Furthermore, the reaction of Oxone® with N,N-diethyl-1-pyrene-carboxamide was applied for fluorescence sensing of this oxidant.24
We found that Oxone® reacts smoothly with thioamides 1a–j in acetonitrile–water at room temperature to afford the corresponding amides 2a–j in high yields (86–100%, Scheme 1).
In order to complete the series for photophysical studies, we also synthesized pyrene amides 2k–m bearing bulky substituents at the nitrogen atom (Scheme 2). Since the isothiocyanates required for syntheses of these compounds via Friedel–Crafts reaction were not available we prepared them from pyrene-1-carboxylic acid and the corresponding amines in the presence of N,N′-dicyclohexylcarbodiimide (DCC). The isolated yields of 2k–m were in the range of 85–90%.
 | ||
| Scheme 2 Synthesis of pyrene-1-carboxamides 2k–m. | ||
All synthesized compounds were fully characterized by spectroscopic methods and elemental analyses.
Solid-state fluorescence of pyrene-1-carboxamides 2a–m
Solution fluorescence of various pyrene-1-carboxamides was recently thoroughly studied by Konishi et al.14–16 During the course of our synthetic work, we discovered that these compounds are also solid-state emitters. Organic solid-state fluorescence is of great interest currently because of its relevance to numerous practical applications, e.g. in organic light-emitting diodes, organic light-emitting field-effect transistors, solid-state lasers and sensors.25–27 It critically depends on interactions of molecular fluorophores (π–π interactions, formation of H- or J-aggregates, excimers, etc.) in powders, crystals or solid films.38We measured solid-state excitation spectra, emission spectra and fluorescence quantum yields of amides 2a–d, g, h, k–m (compound 2j proved non fluorescent). Corresponding spectra are displayed in Fig. 1 and spectroscopic data are gathered in Table 1.
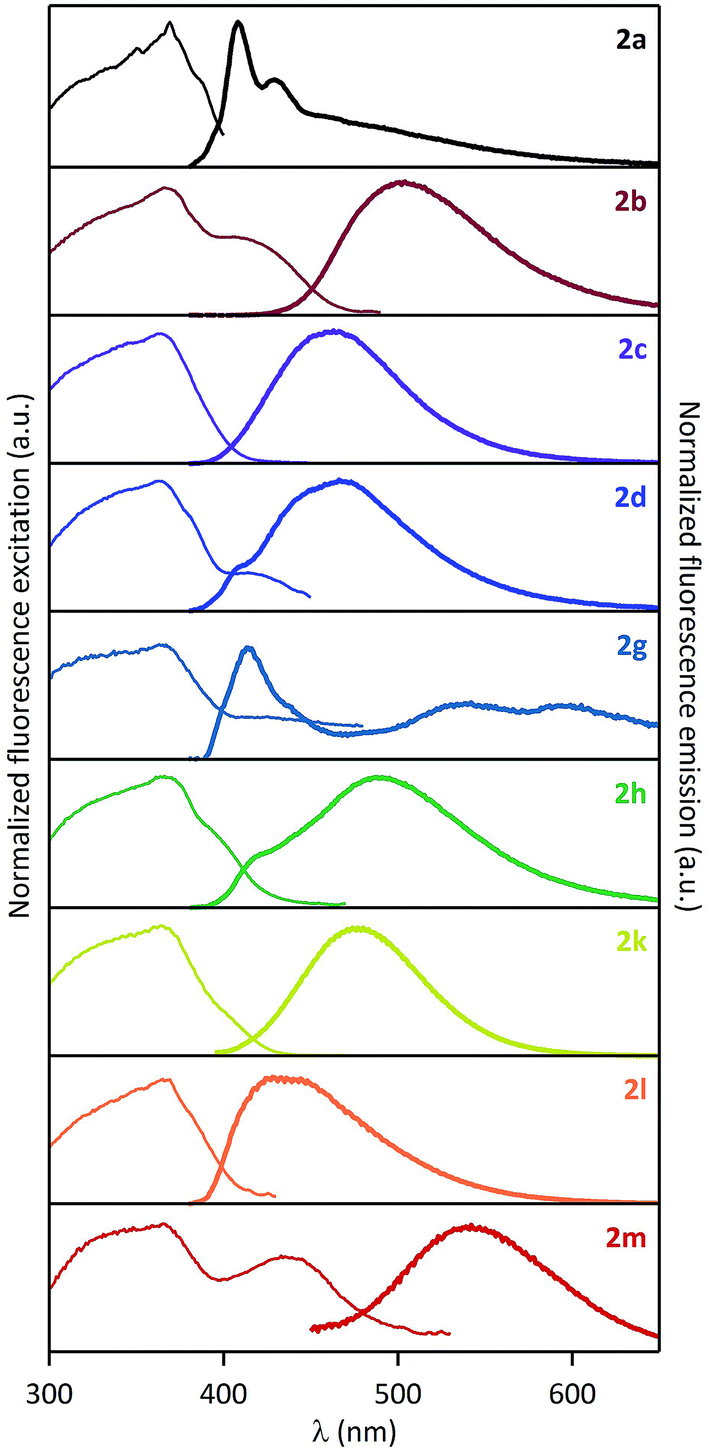 | ||
| Fig. 1 Normalized solid-state excitation (left) and emission (right) spectra of pyrene-1-carboxamides 2a–d, g, h, k–m. | ||
| Amide | Excitationaλmax/nm | Emissionaλmax/nm | Φ F/% |
|---|---|---|---|
| a Shoulders are noted (sh). | |||
| 2a (R = H) | 369 | 408, 430 | 6 |
| 2b (R = iso-Pr) | 367, 410(sh) | 504 | 17 |
| 2c (R = t-Bu) | 364 | 463 | 59 |
| 2d (R = n-hexyl) | 363, 415(sh) | 406(sh), 466 | 12 |
| 2g (R = Cy) | 367 | 414 | 0.2 |
| 2h (R = Bn) | 366, 400(sh) | 416(sh), 488 | 20 |
| 2k (R = 1-adamantyl) | 364 | 477 | 62 |
| 2l (R = CHPh2) | 365 | 428 | 12 |
| 2m (R = CPh3) | 365, 433 | 542 | 7 |
It is visible in Table 1 that substituents at the nitrogen atom strongly influence solid-state emissive properties of the synthesized amides. On one side, the primary amide 2a shows a weak structured violet fluorescence (ΦF = 6%), that is similar in shape to its fluorescence spectrum when dissolved in chloroform (Fig. 2A). However, compared to the solution spectrum, the solid-state emission spectrum of 2a is red-shifted by 25 nm, and a long tail can be observed at wavelengths over 450 nm. This observation suggests that the interaction of fluorophores influencing solid-state fluorescence is in this case rather limited. On the other side, the solid-state excitation/emission spectra of the secondary amides have a wide variety of shapes, with corresponding quantum efficiencies varying from 0.2% to 62%, as a function of the substituents. When analyzing in detail the solid-state emission spectra, we can distinguish several cases: (i) one compound (2g) shows a clear monomer contribution, peaking at 414 nm, associated with a structured shape and a very low fluorescence quantum yield (ΦF = 0.2%); (ii) several compounds (2b–c, 2k–m) display a large red-shifted single band, with quantum yields in the 7–62% range; and (iii) two compounds (2d and 2h) show a well-apparent shoulder at the violet edge of the spectrum (406–416 nm) and a main emission band in the range of 450–600 nm, with intermediate quantum yields (12% and 20%, respectively). The spectral signatures highlighted in the two latter cases are clear indications that the solid-state emission originates from aggregates such as preformed dimers, or excimers. Moreover, the excitation spectra depicted in Fig. 1 all show a main maximum band around 363–369 nm, but also eventually a shoulder (or even a well-defined separated band) at longer wavelengths, in the range of 400–433 nm (amides 2b, 2d, 2h, 2l, see Table 1). This observation supports the presence of aggregates in the ground state. In spite of this variety of fluorescence properties, a general tendency can be drawn i.e. that: the increase of the size of the alkyl substituent at the nitrogen atom brings about an increase in the fluorescence quantum yield. Indeed, the amides 2c and 2j bearing a bulky N-tert-butyl and 1-adamantyl groups are the most efficient solid-state emitters of the series (ΦF = 59% and 62%, respectively). On the other hand, N-phenyl substitution (compound 2g) leads practically to disappearance of the solid-state emissive properties. The N-benzyl amide 2h shows moderate fluorescence quantum yield (20%) and the attachment of the second and third phenyl ring to the sp3 carbon (amides 2l and 2m, respectively) brings about decrease of the fluorescence quantum yield.
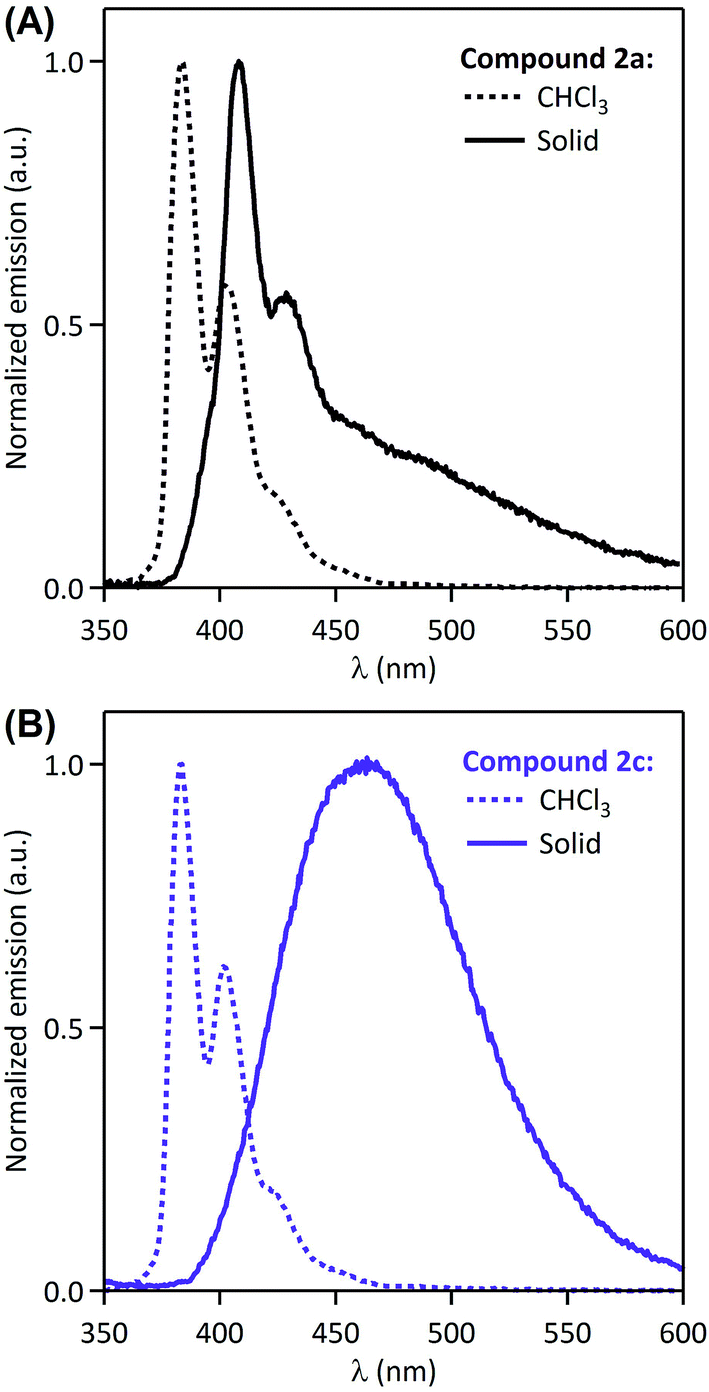 | ||
| Fig. 2 Normalized solution (CHCl3) and solid-state fluorescence emission spectra of 2a (A) and 2c (B). | ||
Practically, the colors of solid-state fluorescence emitted by the investigated series of amides are shown in the CIE diagram (Fig. 3). As a consequence of the different fluorescence spectral shapes of these pyrene derivatives, the solid-state emission expands over a wide range of colors, from violet-blue to green and even yellow, depending on the substituents.
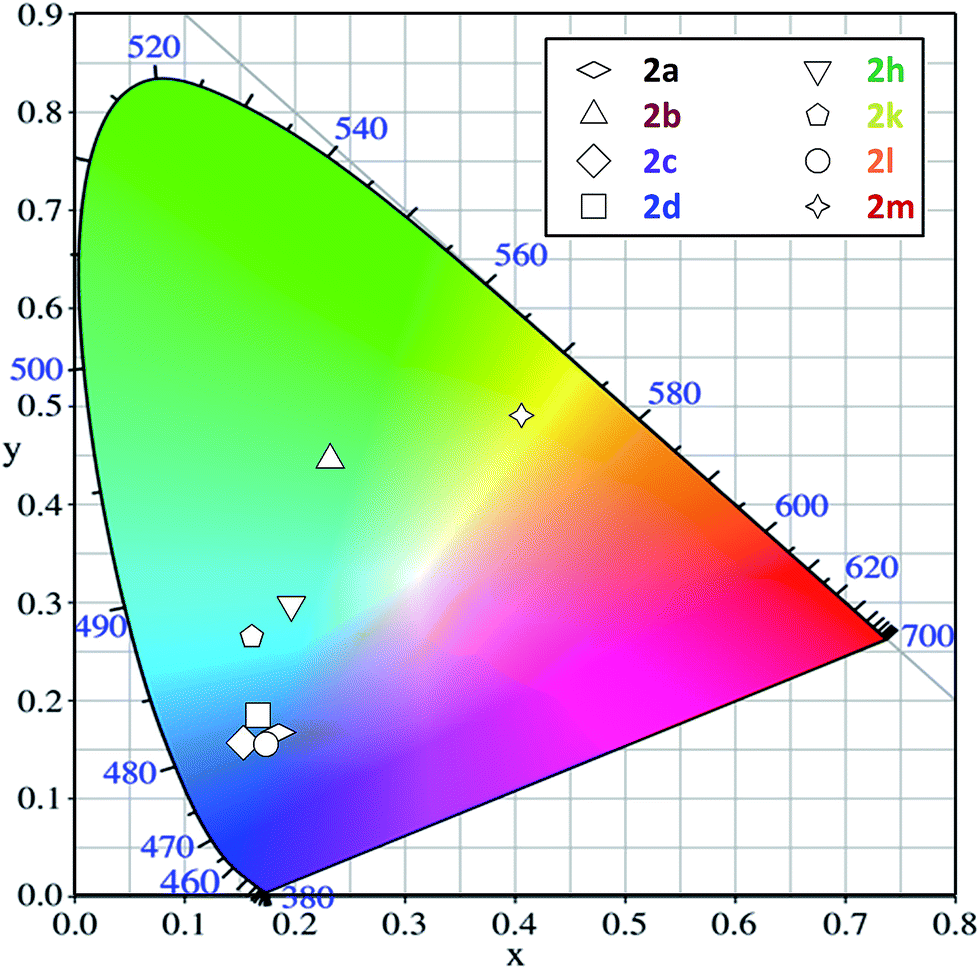 | ||
| Fig. 3 CIE chromacity diagram for compounds 2a–d, h, k–m. | ||
It is worth noting that the most efficient compounds (2c and 2j) emit in the blue region. Interestingly, the former compound has co-ordinates close to those required for deep-blue emission (y < 0.15; x + y < 0.30, cf. ESI†).39 The substituents at the nitrogen atom shift the fluorescence into the blue-green (compound 2b) or even the green-yellow region (compound 2l). However, this bathochromic shift is accompanied by a decrease in emission efficiency.
In order to deepen our investigation of the photophysical properties of pyrene carboxamide derivatives in the solid-state, we recorded fluorescence decays for the whole series 2a–d, g–h, k–m at variable emission wavelengths. The time-correlated single photon counting (TCSPC) method was employed for short decays (decay time constants < 35 ns), whereas the laser flash fluorometry was used for longer decays (including decay time constants > 35 ns). The Fig. 4 displays the solid-state fluorescence decay curves of the compounds 2a–d, g–h, k–m recorded using these experimental methods, at their respective maximum emission wavelengths. In all cases, the fluorescence decays reveal a multiexponential behavior, which reflects the complexity of these systems in the solid-state. They were satisfactorily fitted with a sum of two, three or four exponentials, and the corresponding parameters are compiled in the Table 2. As was mentioned in the previous paragraph, the derivatives 2a and 2g mostly show monomer emission, and constitute a first group of compounds. This translates into relatively short fluorescence decays, with time constants in the range of a few nanoseconds for 2a, and even in the range of several hundreds of picoseconds for 2g. Thus, the different time constants may represent different populations of monomers in the solid. The time-resolved fluorescence investigations reveal a very specific situation for compounds 2b, 2d and 2h (the second group of compounds). Indeed, they show a strong emission wavelength dependence of their fluorescence decays, as is highlighted in Fig. 5, with a multiexponential decay at short emission wavelength (recorded at the blue edge of the emission spectrum), and a short rise time followed by multiexponential decay at long emission wavelength (recorded at the red edge of the emission spectrum). A global analysis was carried out on these data sets (see Table 2), which shows that the short rise time measured at long emission wavelength (0.3–1.6 ns) has the same order of magnitude as the shorter decay time recorded at short emission wavelength (0.5–0.7 ns). This finding may be consistent with the dynamics of fast excimer formation in the solid-state, and is rather compatible with the spectral features described in the previous paragraph. In this frame the longest decay time constant, determined for 2b, 2d and 2h to be 23.7 ns, 24.8 ns, and 34.1 ns, respectively, corresponds specifically to excimer emission, since its relative fraction of intensity increases at longer emission wavelength (Table 2). Then the last group of four compounds, namely 2c and 2j–l, exhibits intermediate behavior between purely monomeric emission (such as 2a and 2g) and well-defined excimer formation (such as 2b, 2d and 2h). On the one hand, the 2c and 2k–m derivatives do not show any rise time at long wavelength emission, which excludes the excimer formation mechanism in these cases. On the other hand, their fluorescence decays are rather long and their emission spectra are broad, red-shifted, without any vibrational structure, which is inconsistent with simple monomer emission. We could then assume that, for 2c and 2k–m, solid-state fluorescence arises from strongly emissive aggregates or dimers preformed in the ground state. Several populations of emitters may exist in the solid state, as is suggested by the multiexponential decays recorded for these compounds. It is worth noting that the situation of compounds 2c and 2k is quite unique, with extremely long decay components (τ1 ∼ 10 ns and τ2 ∼ 45 ns for 2c; τ1 ∼ 12 ns and τ2 ∼ 70 ns for 2k) associated with unprecedented high quantum yields (59% and 62%, respectively). Therefore, for these two derivatives we expect that the formation of aggregates in the crystalline structure and the specific π–π interaction between pyrenyl groups are extremely favorable, thus leading to such effective solid-state fluorescence.
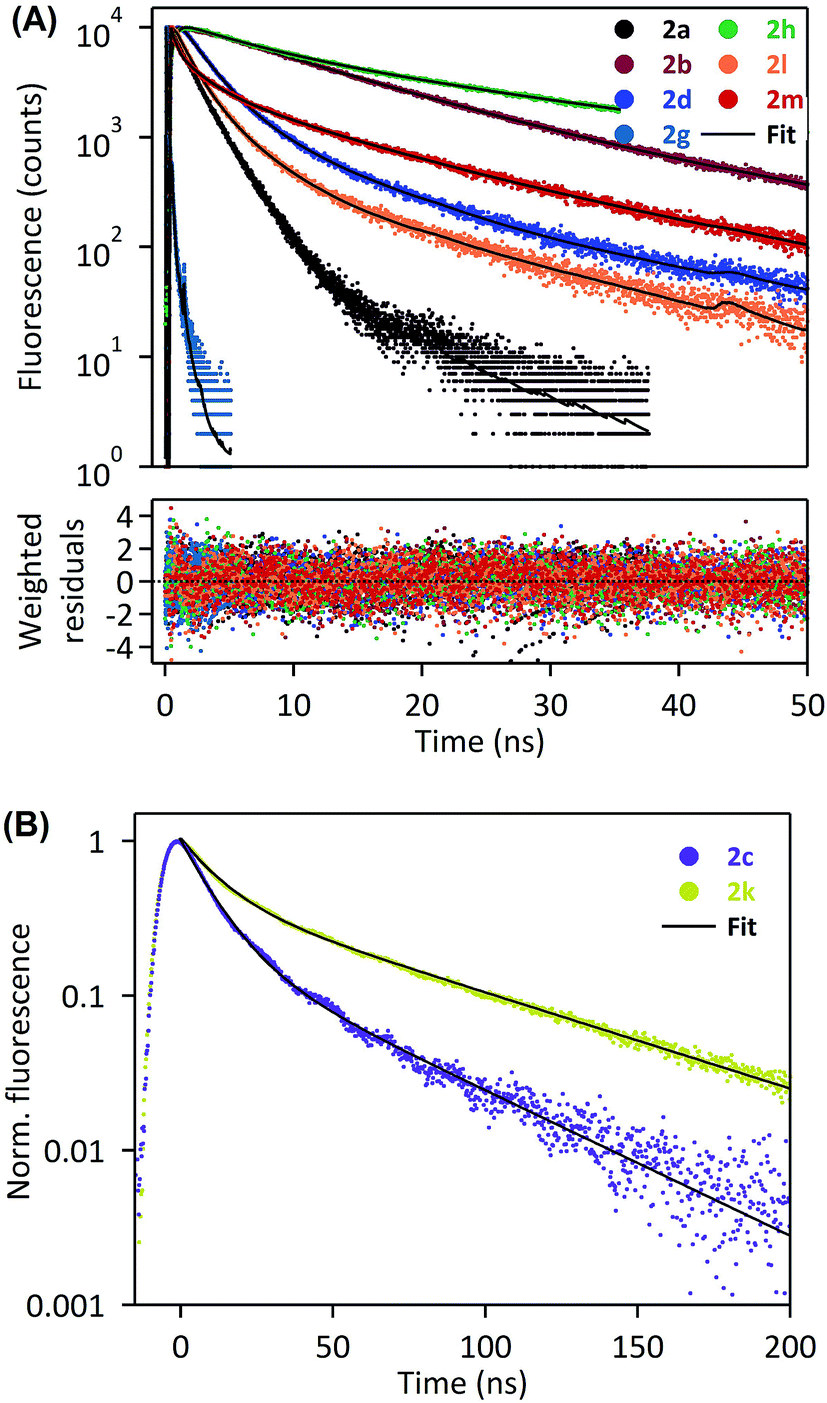 | ||
| Fig. 4 Fluorescence decays of 2a, b, d, g, h, l, m in the solid state, recorded by the TCSPC system at λexc = 370 nm (A), or laser flash fluorometry at λexc = 355 nm (B). | ||
| λ em a (nm) | τ 1 (ns) | τ 2 (ns) | τ 3 (ns) | τ 4 (ns) | A 1 (f1b) | A 2 (f2b) | A 3 (f3b) | A 4 (f4b) | χ r 2 c | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Selected emission wavelength by means of a monochromator with a 50 nm bandwidth. b Intensity fractions were calculated using the following equation: fi = Aiτi/ΣAjτj. c Global χr2, obtained for a global analysis of the full set of fluorescence decay curves, calculated for TCSPC experiments. d Recorded by the TCSPC system, λexc = 370 nm. e Recorded by the laser flash photolysis spectrometer, λexc = 355 nm. | ||||||||||
| 2a d | 410 | 1.3 | 2.3 | 10.4 | — | 0.47 (0.32) | 0.52 (0.65) | 0.01 (0.03) | — | 1.13 |
| 2b d | 460 | 0.7 | 8.8 | 23.7 | — | 0.20 (0.01) | 0.65 (0.60) | 0.15 (0.38) | — | 1.20 |
| 505 | 1.6 | 9.9 | 23.7 | −0.21 (−0.02) | 0.97 (0.64) | 0.25 (0.39) | 1.05 | |||
| 570 | 1.6 | 10.6 | 23.7 | −0.45 (−0.04) | 1.06 (0.57) | 0.39 (0.47) | 1.15 | |||
| 2c e | 420 | 9 | 42 | — | — | 0.85 (0.54) | 0.15 (0.46) | — | — | — |
| 465 | 10 | 46 | 0.81 (0.48) | 0.19 (0.52) | ||||||
| 520 | 11 | 45 | 0.77 (0.46) | 0.23 (0.54) | ||||||
| 2d d | 410 | 0.7 | 2.2 | 7.2 | 24.8 | 0.61 (0.28) | 0.34 (0.47) | 0.05 (0.22) | 0.01 (0.03) | 1.02 |
| 465 | 0.3 | 2.2 | 7.2 | 24.8 | −1.60 (−0.06) | 2.17 (0.57) | 0.38 (0.34) | 0.05 (0.15) | 1.11 | |
| 520 | 0.3 | 2.2 | 7.2 | 24.8 | −2.92 (−0.06) | 3.05 (0.46) | 0.73 (0.36) | 0.15 (0.25) | 1.08 | |
| 2g d | 415 | 0.15 | 0.7 | — | — | 0.98 (0.89) | 0.02 (0.11) | — | — | 1.08 |
| 2h d | 420 | 0.5 | 2.1 | 9.3 | 34.1 | 0.54 (0.14) | 0.38 (0.39) | 0.08 (0.35) | 0.01 (0.13) | 1.08 |
| 490 | 0.5 | 2.1 | 9.3 | 34.1 | −1.14 (−0.02) | 0.28 (0.02) | 1.08 (0.28) | 0.78 (0.72) | 1.18 | |
| 550 | 0.5 | 2.1 | 9.3 | 34.1 | −2.23 (−0.02) | 0.29 (0.01) | 1.64 (0.26) | 1.30 (0.75) | 1.20 | |
| 2k e | 440 | 9 | 68 | — | — | 0.80 (0.35) | 0.20 (0.65) | — | — | — |
| 480 | 12 | 70 | 0.58 (0.19) | 0.42 (0.81) | ||||||
| 540 | 14 | 77 | 0.54 (0.18) | 0.46 (0.82) | ||||||
| 2l d | 430 | 1.3 | 3.4 | 14.4 | — | 0.66 (0.36) | 0.30 (0.41) | 0.04 (0.24) | — | 1.10 |
| 2m d | 540 | 0.3 | 1.8 | 7.7 | 19.6 | 0.44 (0.03) | 0.27 (0.12) | 0.18 (0.35) | 0.10 (0.49) | 1.09 |
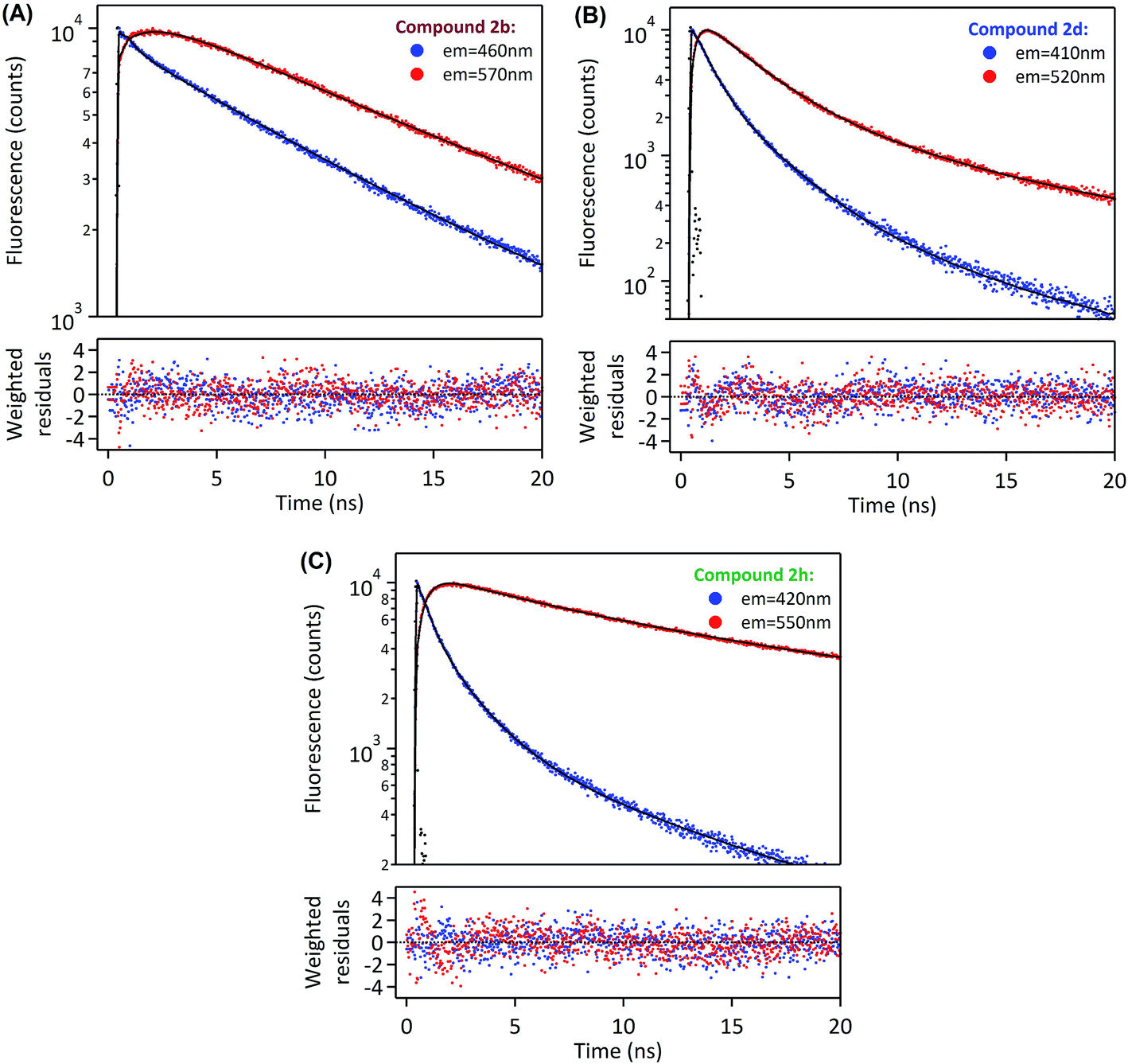 | ||
| Fig. 5 Fluorescence decays of (A) 2b, (B) 2d, (C) 2h in the solid state, recorded by the TCSPC system at two different emission wavelengths: blue edge of the emission spectrum (blue dots) and red edge of the emission spectrum (red dots) (λexc = 370 nm). | ||
Solid-state excimer emission is a relatively common phenomenon for polycyclic aromatic compounds (including pyrene).40–42
X-ray diffraction study of 2c
We intended to compare molecular packings in the crystals of the synthesized amides but we were able to obtain crystals suitable for X-ray diffraction analysis only for 2c. The compound crystallized in the space group P21/c of the monoclinic system after a long period of standing (2–3 weeks) in a concentrated solution in DMSO in an open tube (presumably moisture slowly diffused into the solution and decreased the solubility of 2c). The molecular structure of 2c is shown in Fig. 6.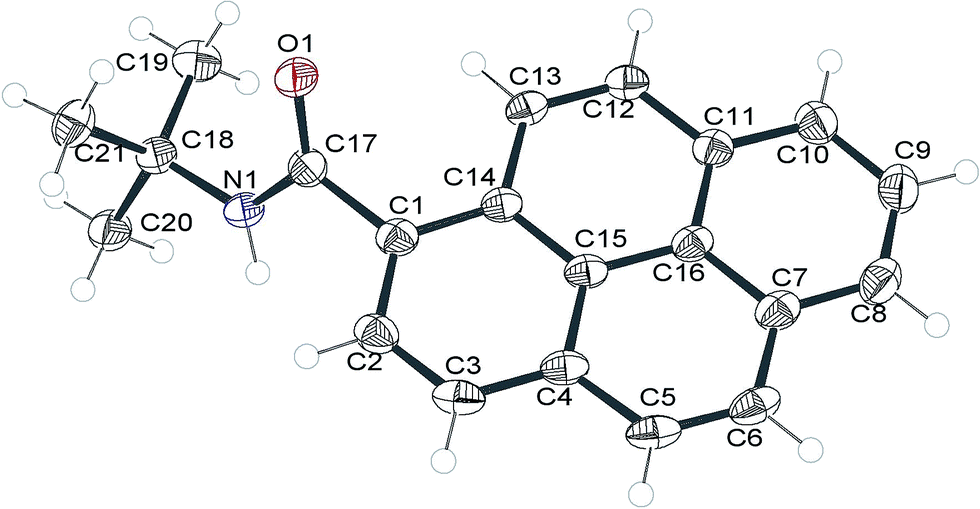 | ||
| Fig. 6 Molecular structure of 2c (ORTEP representation). Displacement ellipsoids were drawn at 50% probability level. | ||
The pyrenyl moiety is slightly bent, with the angle between the planes of rings C1–C2–C3–C4–C15–C14 and C7–C8–C9–C10–C11–C16 ∼4°. The amide group is twisted by 60.6(2) degrees from the average plane of the pyrenyl moiety. Together with the C1–C17 bond length greater than 1.5 Å this suggests that there is only a very weak electronic conjugation between these units.
The crystallographic structure reveals that the molecules of 2c form π-stacked dimers with short C⋯C distances (Fig. 7), thus suggesting strong electronic interactions between the two pyrenyl moieties, which is in very good agreement with the photophysical investigations that were described and discussed in the previous section. This strong π–π interaction in the ground state seems to be at the origin of the very high solid-state fluorescence quantum yield of the 2c derivative, which is associated with its very slow fluorescence decay.
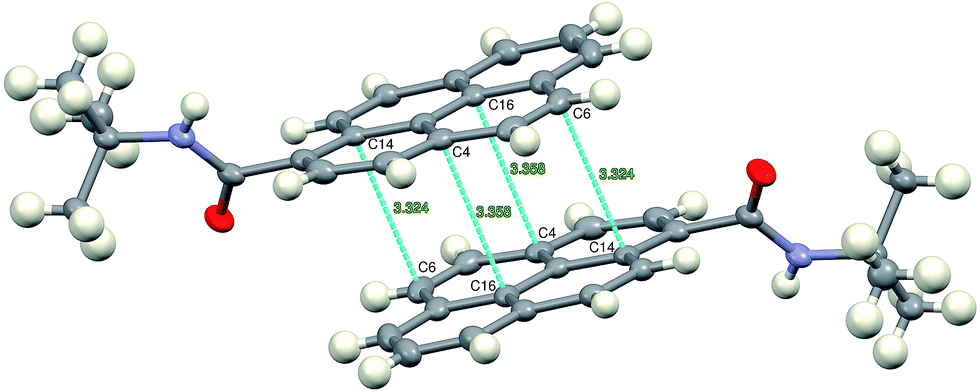 | ||
| Fig. 7 Dimers of π-stacked molecules of 2c, related by the crystallographic center of symmetry. | ||
The intermolecular N1 – H1⋯O1 hydrogen bonds formed between the molecules packed along the crystallographic c direction bring about the formation of infinite chains of H-bonded molecules, with the N⋯O vector almost parallel to c direction and adjacent pyrenyl moieties in the crystal structure located at an angle of 63.0(3) degrees (Fig. 8).
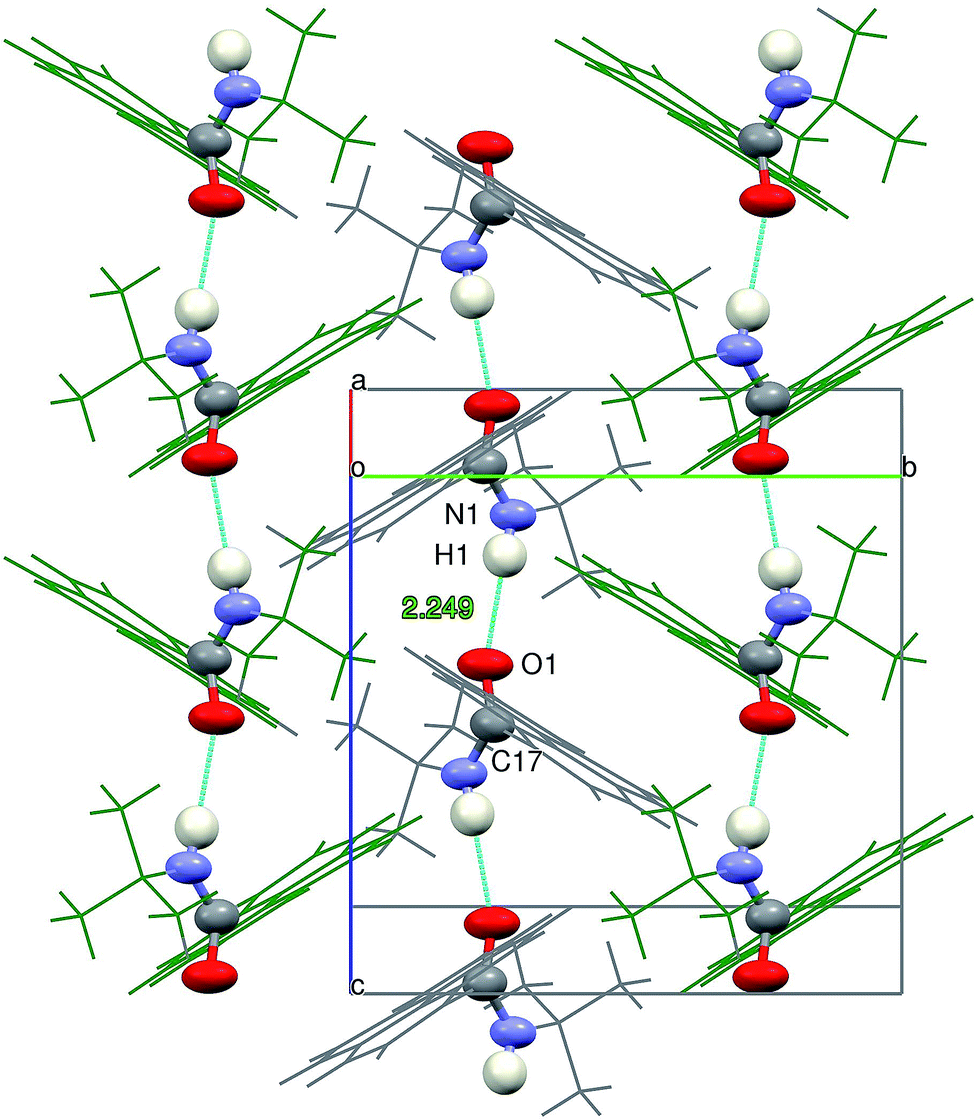 | ||
| Fig. 8 View of the N–H⋯O hydrogen bond in the context of crystal packing of 2c along a* direction. The atoms directly involved in the H-bond network are represented with thermal ellipsoids at 90% probability level, to enhance the pattern. The remaining atoms are represented as wires in gray or in green in order to differentiate between the adjacent π⋯π interacting chains of H-bonded atoms. | ||
Crystal packing viewed along b direction confirms the formation of columns of π-stacked dimers of 2c. Along the crystallographic a direction the crystal structure is stabilized only by van-der Waals interactions of the tert-butyl moieties (Fig. 9).
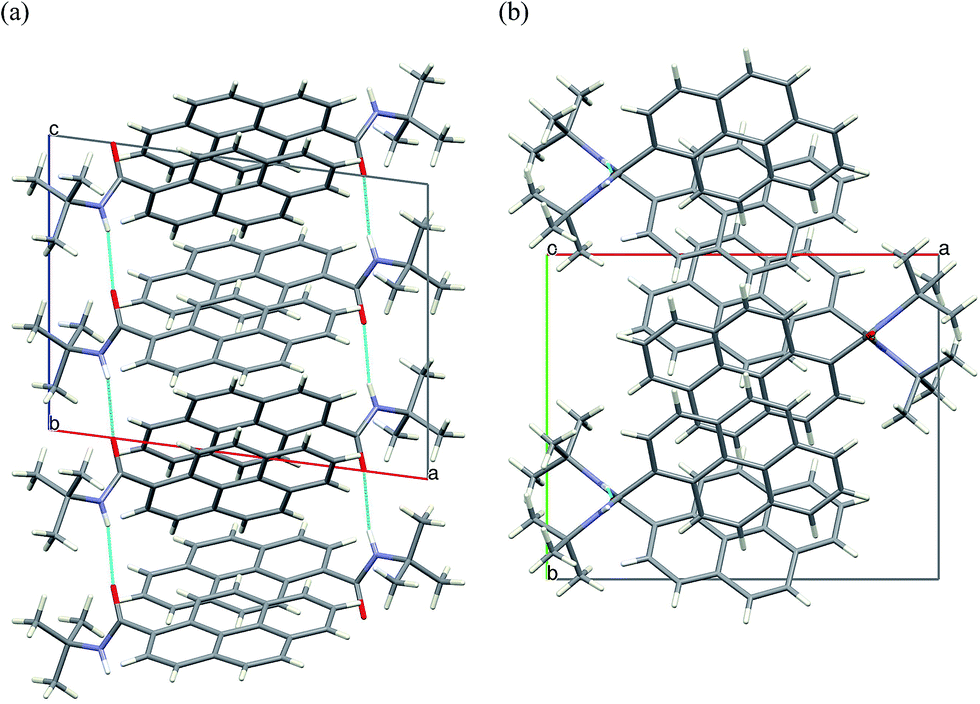 | ||
| Fig. 9 Crystal packing of 2c viewed along the crystallographic (a) b-axis (b) c-axis. H-bonds marked in cyan. | ||
Conclusion
We have developed a simple and efficient synthetic route to primary and secondary pyrene-1-carbothioamides and carboxamides. The amides emit fluorescence in the solid state with quantum efficiencies up to ∼60%. The primary amide in the solid state mainly displays structured monomer emission, whereas several secondary amides show a dynamic excimer formation leading to red-shifted emission. The strong solid-state fluorescence of the most efficient fluorophores, N-tert-butyl- and N-(adamant-1-yl)pyrene-1-carboxamide can be attributed to preformed dimers or aggregates in the ground state, associated with very long decay times.Experimental
Solvents were purified before use by reported methods. All reagents were purchased from Sigma-Aldrich and used without further purification. 1H and 13C NMR spectra were recorded in CDCl3 (if not indicated) or in DMSO-d6 on a Bruker ARX 600 MHz (600 MHz for 1H and 151 MHz for 13C). Chemical shifts were referenced relative to solvent signals. Spectra were recorded at room temperature (291 K), chemical shifts are in ppm and coupling constants in Hz. IR spectra were run on an FT-IR Nexus spectrometer. Column chromatography was carried out on silica gel 60 (0.040–0.063 mm, 230–400 mesh, Fluka). Elemental analyses were performed at the Laboratory of Microanalysis in the Centre of Molecular and Macromolecular Studies in Łódź, Poland.Pyrene-1-carbothioamides 1a–j
1a . Yellow powder (165 mg, 63%), m.p. 179–180 °C; 1H NMR: δ 10.40 (s, 1H), 9.93 (s, 1H), 8.39 (d, J = 9 Hz, 1H), 8.32 (t, J = 6.6 Hz, 2H), 8.28 (d, J = 6.6 Hz, 1H), 8.25 (d, J = 9 Hz, 1H), 8.20 (q, J = 9 Hz, 2H), 8.10 (t, J = 7.8 Hz, 1H), 8.02 (d, J = 7.8 Hz, 1H); 13C NMR: δ 202.76, 138.82, 130.77, 130.64, 130.22, 127.75, 127.11, 126.48, 125.60, 125.29, 125.18, 124.30, 124.15, 123.71, 123.60; IR (cm−1) 3396, 3242, 3047, 1615, 1397, 1291, 914, 846 cm−1; anal. calcd for C17H11NS: C, 78.13; H, 4.24; N, 5.36; S, 12.27; found: C, 78.15; H, 4.22; N, 5.41%.
1b . Yellow powder (291 mg, 96%), m.p. 186.5–187 °C; 1H NMR: δ 8.25–8.26 (m, 1H), 8.18 (t, J = 9.6 Hz, 2H), 8.05–8.09 (m, 3H), 7.99–8.02 (m, 3H), 7.52 (s, 1H), 4.98–5.04 (m, 1H), 1.43 (d, J = 6.8 Hz, 6H), 13C NMR: δ 199.33, 138.72, 131.64, 131.23, 130.65, 128.53, 128.12, 127.10, 127.08, 126.26, 126.12, 125.73, 125.43, 124.64, 124.55, 124.51, 124.49, 123.49, 47.93, 21.37; IR (KBr) 3267, 3041, 2968, 2926, 1523, 1394, 849 cm−1; anal. calcd for C20H17NS: C, 79.17; H, 5.65; N, 4.62; S, 10.57; found: C, 79.15; H, 5.69; N, 4.66%.
1c . Yellow powder (292 mg, 93%), m.p.: 184–185 °C; 1H NMR: δ 8.30 (d, J = 9 Hz, 1H), 8.19 (t, J = 7.2 Hz, 2H), 8.13 (dd, J1 = 2.4 Hz, J2 = 9.0 Hz, 2H), 8.08 (d, J = 9 Hz, 1H), 8.03 (dd, J1 = 2.4 Hz, J2 = 7.8 Hz, 2H), 8.01 (d, J = 9 Hz, 1H), 7.48 (s,1H), 1.79 (s, 9H); 13C NMR: δ 200.23, 140.54, 131.44, 131.32, 130.74, 129.53, 128.59, 128.02, 127.16, 126.28, 125.79, 125.72, 125.39, 124.75, 124.64, 124.32, 123.54, 56.83, 27.87; IR (KBr): 3355, 3039, 2966, 1512, 1400, 1362, 843 cm−1; anal. calcd for C21H19NS: C, 79.45; H, 6.03; N, 4.41; S, 10.10; found: C, 79.39; H, 6.07; S, 10.04%.
1d . Yellow powder (314 mg, 91%), m.p. 156–157 °C; 1H NMR: δ 8.27 (d, J = 9 Hz, 1H), 8.20 (d, J = 7.8 Hz, 1H), 8.19 (d, J = 7.2 Hz, 1H), 8.07–8.11 (m, 3H), 8.01–8.03 (m, 3H), 7.62 (s, 1H), 3.96 (t, J = 7.2 Hz, 1H), 3.95 (t, J = 7.2 Hz, 1H), 1.8 (kwintet, J = 7.4 Hz, 2H), 1.44–1.48 (m, 2H), 1.34–1.38 (m, 4H), 0.93 (t, J = 7.2 Hz, 3H); 13C NMR: δ 200.70, 138.76, 131.74, 131.29, 130.72, 128.53, 128.21, 127.14, 126.31, 126.29, 125.78, 125.47, 124.69, 124.65, 124.57, 124.53, 123.71, 46.60, 31.42, 28.09, 26.78, 22.53, 13.96; IR (KBr) 3169, 3036, 2930, 1545, 1397, 1071, 847 cm−1. Anal. calcd for C23H23NS: C, 79.96; H, 6.71; N, 4.05; S, 9.28; found: C, 79.88; H, 6.70; N, 4.16%.
1e . Yellow powder (323 mg, 94%), m.p.: 206–207 °C; 1H NMR: δ 10.60 (d, J = 7.2 Hz, 1H), 8.32 (d, J = 9.6 Hz, 1H), 8.30 (d, J = 10.8 Hz, 1H), 8.28 (d, J = 10.8 Hz, 1H), 8.17–8.23 (m, 4H), 8.10 (t, J = 7.8 Hz, 1H), 7.93 (d, J = 7.8 Hz, 1H), 4.62–4.64 (m, 1H), 2.17 (d, J = 9.6 Hz, 2H), 1.83 (d, J = 12.6 Hz, 2H), 1.67 (d, J = 13.2 Hz, 1H), 1.38–1.48 (m, 4H), 1.19–1.23 (m, 1H), 13C NMR: δ 196.64, 139.09, 130.76, 130.47, 130.23, 127.81, 127.62, 127.11, 126.45, 125.55, 125.23, 124.46, 124.31, 123.99, 123.72, 123.63, 54.52, 30.60, 25.06, 24.56; IR (KBr) 3346, 2928, 2856, 1519, 1396, 845 cm−1; anal. calcd for C23H21NS: C, 80.42; H, 6.16; N, 4.08; S, 9.34; found: C, 80.39; H, 6.22; N, 4.16%.
1f . Yellow oil (285 mg, 78%) 1H NMR: δ 13.25 (s, 1H), 8.34 (d, J = 7.8 Hz, 1H), 8.32 (d, J = 7.8 Hz, 1H), 8.29 (d, J = 7.8 Hz, 1H), 8.28 (d, J = 9.6 Hz, 1H), 8.24 (d, J = 9.0 Hz, 1H), 8.21 (d, J = 9.0 Hz, 1H), 8.19 (d, J = 7.2 Hz, 1H), 8.10 (d, J = 7.2 Hz, 1H), 8.07 (d, J = 7.8 Hz, 1H), 7.97 (d, J = 7.8 Hz, 2H), 7.66 (t, J = 7.2 Hz, 1H), 7.52 (t, J = 7.8 Hz, 2H); 13C NMR: δ 197.49, 165.11, 139.58, 137.97, 133.50, 133.31, 132.03, 130.99, 130.76, 130.08, 129.98, 129.06, 128.75, 128.44, 127.96, 127.18, 126.47, 125.85, 125.14, 124.97, 124.34, 123.35; IR (KBr) 1713, 1698, 1598, 1470, 1170, 846 cm−1; anal. calcd for C24H15NOS: C, 78.88; H, 4.14; N, 3.83; O, 4.38; S, 8.77; found: C, 79.03; H, 4.02; N, 3.97%.
1g . Yellow powder (287 mg, 85%) 1H NMR: δ 12.32 (s, 1H), 8.35 (d, J = 7.8 Hz, 1H), 8.34 (d, J = 7.8 Hz, 1H), 8.33 (d, J = 7.8 Hz, 2H), 8.26 (d, J = 9.0 Hz, 1H), 8.24 (d, J = 3.6 Hz, 2H), 8.12 (d, J = 7.8 Hz, 2H), 8.10 (d, J = 7.8 Hz, 2H), 7.52 (t, J = 7.8 Hz, 2H), 7.35 (t, J = 7.2 Hz, 1H); 13C NMR: δ 197.96, 139.73, 139.64, 130.79, 130.23, 128.62, 128.45, 128.15, 127.85, 127.13, 126.53, 126.38, 125.73, 125.57, 125.39, 124.70, 124.47, 123.94, 123.74, 123.68, 123.27, 123.24; IR (KBr) 3329, 3037, 1594, 1497, 1356, 846 cm−1; anal. calcd for C23H15NS: C, 81.87; H, 4.48; N, 4.15; S, 9.50; found: C, 81.84; H, 4.51; N, 4.26%.
1h . Yellow powder (302 mg, 86%), m.p.: 148–149 °C, 1H NMR: δ 8.32 (d, J = 9.0 Hz, 1H), 8.19 (d, J = 12.2 Hz, 1H), 8.18 (d, J = 12.2 Hz, 1H), 8.10 (t, J = 7.8 Hz, 1H), 8.08 (d, J = 10.2 Hz, 1H), 8.06 (t, J = 7.8 Hz, 2H), 8.02 (t, J = 7.8 Hz, 1H), 8.01 (d, J = 9.0 Hz, 1H), 7.84 (s, 1H), 8.02 (d, J = 7.8 Hz, 2H), 7.38–7.41 (m, 2H), 7.33–7.36 (m, 1H), 5.17 (d, J = 5.4 Hz, 2H); 13C NMR: δ 200.81, 138.34, 136.04, 131.88, 131.28, 130.68, 129.06, 128.86, 128.63, 128.44, 128.32, 128.28, 127.63, 127.13, 126.43, 126.34, 125.83, 125.53, 124.71, 124.68, 124.53, 123.66, 50.81; IR (KBr) 3211, 3083, 3028, 1522, 1337, 1229, 948, 846 cm−1; anal. calcd for C24H17NS: C, 82.02; H, 4.88; N, 3.99; S, 9.12; found: C, 82.07; H, 4.85; N, 4.14%.
1i . Yellow powder (323 mg, 88%), m.p.: 189–190 °C; 1H NMR: δ 12.19 (s, 1H), 8.32–8.36 (m, 3H), 8.30 (d, J = 9.0 Hz, 1H), 8.26 (d, J = 9.0 Hz, 1H), 8.22–8.24 (m, 2H), 8.11 (t, J = 7.8 Hz, 2H), 8.00 (d, J = 9.0 Hz, 2H), 7.06 (d, J = 9.0 Hz, 2H), 3.83 (s, 3H); 13C NMR: δ 196.87, 157.35, 139.70, 132.77, 130.79, 130.69, 130.24, 128.07, 127.79, 127.13, 126.51, 125.69, 125.60, 125.35, 124.89, 124.73, 124.69, 124.45, 124.02, 123.75, 123.69, 113.71, 55.30; IR (KBr) 3155, 3049, 2961, 1594, 1509, 1247, 849 cm−1; anal. calcd for C24H17NOS: C, 78.45; H, 4.66; N, 3.81; O, 4.35; S, 8.73; found: C, 78.37; H, 4.72; N, 3.95%.
1j . Yellow oil (332 mg, 87%), 1H NMR: δ 12.77 (s, 1H), 8.47 (d, J = 8.4 Hz, 2H), 8.36–8.40 (m, 4H), 8.34 (d, J = 7.8 Hz, 1H), 8.24–8.28 (m, 4H), 8.15 (d, J = 7.8 Hz, 1H), 8.13 (t, J = 7.2 Hz, 1H); 13C NMR: δ 199.86, 145.19, 144.27, 139.49, 131.07, 130.76, 130.16, 128.42, 128.11, 127.10, 126.62, 125.91, 125.62, 125.55, 124.87, 124.45, 123.73, 123.65, 123.62, 122.89; IR (KBr) 3299, 1595, 1551, 1498, 1332, 1317, 1303, 846, cm−1; anal. calcd for C23H14N2O2S: C, 72.23; H, 3.69; N, 7.33; O, 8.37; S, 8.38; found: C, 72.31; H, 3.59; N, 7.45%.
Pyrene-1-carboxamides 2a–i
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 → 99:1).
:0 → 99:1).
2a . White powder (210 mg, 86%), m.p.: 257 - 258 °C; 1H NMR (DMSO-d6): δ 8.63 (d, J = 9.6 Hz, 1H), 8.34 (t, J = 9.0 Hz, 2H), 8.32 (d, J = 7.8 Hz, 1H), 8.30 (d, J = 7.8 Hz, 2H), 8.22 (d, J = 9.0 Hz, 1H), 8.18 (d, J = 9.0 Hz, 1H), 8.16 (s, 1H), 8.11 (t, J = 7.8 Hz, 1H), 7.73 (s, 1H); 13C NMR (DMSO-d6): δ 170.86, 131.76, 131.55, 130.66, 130.13, 128.17, 127.93, 127.72, 127.14, 126.46, 125.68, 125.48, 125.18, 124.80, 124.30, 123.81, 123.63; IR (KBr) 3339, 3169, 1660, 1622, 1399, 853 cm−1; anal. calcd for C17H11NO: C, 83.25; H, 4.52; N, 5.71; O, 6.52; found: C, 83.38; H, 4.67; N, 5.53%.
2b . White powder (273 mg, 95%), m.p.: 211.5–212.5 °C; 1H NMR (DMSO-d6): δ 8.55 (d, J = 7.8 Hz, 1H), 8.46 (d, J = 9.6 Hz, 1H), 8.34 (d, J = 6.6 Hz, 2H), 8.31 (d, J = 7.8 Hz, 1H), 8.25 (d, J = 9.0 Hz, 1H), 8.24 (d, J = 9.0 Hz, 1H), 8.21 (d, J = 9.0 Hz, 1H), 8.10 (t, J = 9.0 Hz, 2H), 4.26–4.31 (m, 1H), 1.28 (d, J = 6.6 Hz, 6H); 13C NMR (DMSO-d6): δ 167.91, 132.45, 131.32, 130.69, 130.17, 128.06, 127.94, 127.63, 127.15, 126.46, 125.64, 125.45, 125.10, 124.58, 124.29, 123.73, 123.64, 41.13, 22.35; IR (KBr) 3278, 3047, 2975, 1326, 1545, 1530, 1285, 846 cm−1; anal. calcd for C20H17NO: C, 83.59; H, 5.96; N, 4.87; O, 5.57; found: C, 83.49; H, 6.07; N, 4.93%.
2c . White powder (280 mg, 93%), m.p.: 232–233 °C; 1H NMR: δ 8.53 (d, J = 9 Hz, 1H), 8.22 (dd, J1 = 2.4 Hz, J2 = 10.0 Hz, 2H), 8.14 (t, J = 7.8 Hz, 2H), 8.10 (d, J = 9 Hz, 1H), 8.05 (d, J = 7.8 Hz, 1H), 8.03 (t, J = 7.2 Hz, 2H), 5.95 (s, 1H), 1.61 (s, 9H); 13C NMR: δ 169.57, 132.68, 132.21, 131.23, 130.78, 128.78, 128.55, 128.41, 128.27, 127.15, 126.27, 125.69, 125.60, 124.80, 124.53, 124.38, 124.32, 52.29, 29.05; IR (KBr) 3325, 3046, 2977, 2958, 1639, 1523, 1298, 1223, 844 cm−1; anal. calcd for C21H19NO: C, 83.69; H, 6.35; N, 4.65; O, 5.31; found: C, 83.76; H, 6.39; N, 4.53%.
2d . White powder (316 mg, 96%); 1H NMR: δ 8.46 (d, J = 9.6 Hz, 1H), 8.16 (d, J = 7.8 Hz, 2H), 8.03–8.05 (m, 2H), 7.99–8.02 (m, 2H), 7.93–7.96 (m, 2H), 6.29 (s, 1H), 3.54 (q, J = 7.2 Hz, 2H), 1.67 (kwintet, J = 7.2 Hz, 2H), 1.40–1.44 (m, 2H), 1.32–1.36 (m, 4H), 0.92–0.94 (m, 3H); 13C NMR: δ 169.92, 132.24, 131.36, 131.07, 130.63, 128.41, 128.38, 128.34, 126.99, 126.17, 125.64, 125.55, 124.60, 124.36, 124.32, 124.15, 123.70, 40.27, 31.49, 29.68, 26.70, 22.55, 13.99; IR (KBr) 3299, 3037, 2928, 1625, 1536, 848 cm−1; anal. calcd for C23H23NO: C, 83.85; H, 7.04; N, 4.25; O, 4.86; found: C, 83.89; H, 6.99; N, 4.31%.
2e . White powder (298 mg, 91%); m.p.: 236–237 °C; 1H NMR: δ 8.52 (d, J = 9.0 Hz, 1H), 8.19 (d, J = 7.8 Hz, 2H), 8.11 (d, J = 9.0 Hz, 1H), 8.09 (d, J = 7.8 Hz, 1H), 8.08 (d, J = 9.0 Hz, 1H), 8.04 (d, J = 7.8 Hz, 1H), 8.02 (t, J = 7.8 Hz, 1H), 8.01 (d, J = 9.0 Hz, 1H), 6.05 (d, J = 7.8 Hz, 1H), 4.15–4.21 (m, 1H), 2.17–2.19 (m, 2H), 1.80 (dt, J1 = 3.6 Hz, J2 = 13.8 Hz, 2H), 1.69 (dt, J1 = 3.6 Hz, J2 = 13.2 Hz, 1H), 1.50 (qt, J1 = 3.6 Hz, J2 = 12.0 Hz, 2H), 1.33 (qd, J1 = 3.6 Hz, J2 = 12.0 Hz, 2H), 1.23 (qt, J1 = 3.6 Hz, J2 = 12.2 Hz, 1H); 13C NMR: δ 169.10, 132.31, 131.66, 131.15, 130.70, 128.54, 128.44, 128.36, 127.08, 126.31, 126.24, 125.69, 125.60, 124.72, 124.42, 124.34, 124.26, 49.03, 33.28, 25.58, 24.93; IR (KBr) 3274, 3039, 2934, 2916, 1625, 1532, 1306, 848 cm−1; anal. calcd for C23H21NO: C, 84.37; H, 6.46; N, 4.28; O, 4.89; found: C, 84.30; H, 6.52; N, 4.37%.
2f . Yellow oil (311 mg, 89%); 1H NMR: δ 9.56 (s, 1H), δ 88.58 (d, J = 9.0 Hz, 1H), 8.20 (d, J = 7.2 Hz, 2H), 8.11 (d, J = 10.2 Hz, 2H), 8.08 (d, J = 9.0 Hz, 2H), 8.03 (t, J = 7.8 Hz, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.46 (d, J = 7.8 Hz, 2H), 7.39 (t, J = 7.2 Hz, 2H), 7.32 (t, J = 7.8 Hz, 1H), 6.48 (s, 1H), 4.80 (d, J = 5.4 Hz, 2H); 13C NMR: δ 169.77, 138.25, 132.45, 131.07, 130.67, 130.61, 128.79, 128.57, 128.56, 128.53, 128.03, 127.90, 127.59, 126.99, 126.22, 125.72, 125.64, 124.64, 124.39, 124.31, 124.29, 124.13, 44.29; IR (KBr) 3261, 1644, 1595, 1523, 1441, 1331, 845 cm−1; anal. calcd for C24H15NO2: C, 82.50; H, 4.33; N, 4.01; O, 9.16; Found: C, 82.63; H, 4.45; N, 3.92%.
2g . Yellow powder (296 mg, 92%), m.p.: 253–255 °C; 1H NMR: δ 10.71 (s, 1H), 8.48 (d, J = 9.6 Hz, 1H), 8.40 (d, J = 9.0 Hz, 1H), 8.37 (t, J = 7.8 Hz, 2H), 8.30 (d, J = 7.8 Hz, 1H), 8.29 (d, J = 7.8 Hz, 1H), 8.28 (t, J = 9.0 Hz, 2H), 8.14 (t, J = 7.2 Hz, 1H), 7.88 (d, J = 7.8 Hz, 2H), 8.41 (t, J = 7.8 Hz, 2H), 7.16 (t, J = 7.2 Hz, 1H); 13C NMR: δ 167.56, 139.37, 131.84, 131.76, 130.68, 130.15, 128.69, 128.43, 128.39, 127.76, 127.15, 126.60, 125.90, 125.68, 125.30, 124.39, 124.30, 123.75, 123.69, 123.60, 119.86; IR (KBr) 3260, 3038, 1645, 1524, 1441, 1331, 845 cm−1; anal. calcd for C23H15NO: C, 85.96; H, 4.70; N, 4.36; O, 4.98; found: C, 85.88; H, 4.76; N, 4.42%.
2h . Yellow powder (302 mg, 90%), m.p.: 161–162 °C; 1H NMR: δ 8.58 (d, J = 9.0 Hz, 1H), 8.20 (d, J = 7.2 Hz, 2H), 8.11 (d, J = 10.2 Hz, 2H), 8.08 (d, J = 9.0 Hz, 2H), 8.03 (t, J = 7.8 Hz, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.46 (d, J = 7.8 Hz, 2H), 7.39 (t, J = 7.2 Hz, 2H), 7.32 (t, J = 7.8 Hz, 1H), 6.48 (s, 1H), 4.80 (d, J = 5.4 Hz, 2H); 13C NMR: δ 169.77, 138.25, 132.45, 131.07, 130.67, 130.61, 128.79, 128.57, 128.56, 128.53, 128.03, 127.90, 127.59, 126.99, 126.22, 125.72, 125.64, 124.64, 124.39, 124.31, 124.29, 124.13, 44.29; IR (KBr) 3270, 3030, 2920, 1632, 1529, 1290, 848 cm−1; anal. calcd for C24H17NO: C, 85.94; H, 5.11; N, 4.18; O, 4.77; found: C, 85.87; H, 5.09; N, 4.25%.
2i . White powder (316 mg, 90%), m.p.: 251–252 °C; 1H NMR: δ 10.69 (s, 1H), 8.51 (d, J = 9.0 Hz, 1H), 8.37 (d, J = 9.8 Hz, 1H), 8.36 (t, J = 7.8 Hz, 2H), 8.28 (d, J = 9.0 Hz, 1H), 8.27 (d, J = 9.0 Hz, 1H), 8.25 (t, J = 7.8 Hz, 2H), 8.12 (t, J = 7.2 Hz, 1H), 7.83 (d, J = 4.8 Hz, 2H), 6.99 (d, J = 4.8 Hz, 2H), 3.77 (s, 3H); 13C NMR: δ 167.17, 155.59, 132.64, 131.97, 131.70, 130.70, 130.17, 128.36, 128.28, 127.82, 127.18, 126.57, 125.86, 125.64, 125.39, 124.46, 124.40, 123.79, 123.64, 121.50, 113.86, 55.23; IR (KBr) 3323, 3039, 2929, 1643, 1540, 1340, 1234, 848 cm−1; anal. calcd for C24H17NO2: C, 82.03; H, 4.88; N, 3.99; O, 9.11; found: C, 82.05; H, 4.83; N, 3.91%.
2j . Yellow powder (289 mg, 79%), 1H NMR: δ 11.32 (s, 1H), 8.41 (d, J = 8.4 Hz, 1H), 8.40 (d, J = 7.8 Hz, 1H), 8.30–8.34 (m, 2H), 8.27 (d, J = 7.8 Hz, 1H), 8.26 (d, J = 9.0 Hz, 1H), 8.23 (d, J = 9.0 Hz, 1H), 8.13 (t, J = 7.8 Hz, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 8.4 Hz, 2H) 13C NMR: δ 168.15, 142.69, 133.13, 130.52, 129.88, 129.51, 129.21, 128.32, 127.86, 127.12, 126.92, 126.59, 126.35, 124.78, 124.39, 123.64, 123.15, 122.58, 120.64; IR (KBr) 3172, 1612, 1594, 1514, 1336, 1029, 987, 841 cm−1; anal. calcd for C23H14NO3: C, 75.40; H, 3.85; N, 7.65; O, 13.10; found: C, 75.51; H, 3.89; N, 12.89%.
Pyrene-1 carboxamides 2k–m
2k . White powder (321 mg, 85%). 1H NMR: δ 8.33 (d, J = 7.2 Hz, 1H), 8.32 (d, J = 7.8 Hz, 1H), 8.26 (d, J = 7.8 Hz, 1H), 8.22–8.25 (m, 3H), 8.19 (t, J = 7.8 Hz, 2H), 7.89 (d, J = 7.8 Hz, 1H), 1.63–1.83 (m, 6H), 1.24–1.34 (m, 3H), 0.69–1.11 (m, 6H); 13C NMR: δ 167.92, 131.83, 131.10, 130.59, 130.18, 128.02, 127.67, 127.18, 127.05, 126.43, 125.61, 125.44, 124.81, 124.63, 123.84, 123.48, 123.46, 48.81, 30.99, 30.47, 25.46; IR (KBr) 3306, 3039, 2929, 2855, 1644, 1541, 1339, 847 cm−1; anal. calcd for C27H25NO: C, 85.45; H, 6.64; N, 3.69; O, 4.22; found: C, 85.54; H, 6.59; N, 3.76%.
2l . White powder (369 mg, 90%). 1H NMR (DMSO-d6): δ 9.69 (d, J = 8.4 Hz, 1H), 8.36 (d, J = 9.0 Hz, 1H), 8.35 (d, J = 7.2 Hz, 1H), 8.33 (d, J = 7.8 Hz, 2H), 8.26 (d, J = 9.0 Hz, 1H), 8.23 (d, J = 9.0 Hz, 1H), 8.22 (d, J = 9.6 Hz, 1H), 8.17 (d, J = 7.8 Hz, 1H), 8.12 (t, J = 7.2 Hz, 1H), 7.50 (d, J = 7.8 Hz, 4H), 7.41 (t, J = 7.8 Hz, 4H), 7.30 (t, J = 7.2 Hz, 2H), 6.61 (d, J = 9.0 Hz, 1H); 13C NMR (DMSO-d6): δ 168.21, 131.68, 131.52, 130.62, 130.11, 128.36, 128.20, 128.04, 127.80, 127.43, 127.12, 126.96, 126.48, 125.73, 125.52, 125.29, 124.36, 124.25, 123.69, 123.58, 56.55; IR (KBr) 3325, 3036, 2929, 1631, 1572, 848 cm−1; anal. calcd for C30H21NO: C, 87.56; H, 5.14; N, 3.40; O, 3.89; found: C, 87.45; H, 5.23; N, 3.43%.
2m . White powder (421 mg, 86%), 1H NMR (DMSO-d6): δ 8.48 (d, J = 9.0 Hz, 1H), 8.34 (d, J = 7.8 Hz, 1H), 8.32 (d, J = 8.4 Hz, 2H), 8.24–8.27 (m, 3H), 8.19 (d, J = 8.4 Hz, 1H), 8.13 (d, J = 7.8 Hz, 1H), 8.11 (d, J = 8.4 Hz, 1H), 7.71–7.74 (m, 6H), 7.50–7.52 (m, 3H), 7.44–7.46 (m, 6H); 13C NMR (150 MHz, DMSO): δ 173.01, 153.80, 148.66, 131.97, 131.83, 131.25, 130.84, 129.11, 128.50, 128.19, 128.14, 127.96, 127.93, 127.88, 127.33, 127.16, 126.71, 126.57, 126.47, 125.97, 125.87, 124.70, 124.52, 124.51, 123.56, 122.67; IR (KBr) 3329, 2929, 2852, 1628, 1577, 1310, 847, 705 cm−1; anal. calcd for C36H25NO: C, 88.68; H, 5.17; N, 2.87; O, 3.28; found: C, 88.63; H, 5.13; N, 2.99%.
Acknowledgements
Financial support from the National Science Centre Poland (NCN, Grant Harmonia UMO-2012/04/M/ST5/00712) is gratefully acknowledged.References
- T. M. Figueira-Duarte and K. Muellen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed.
- G. Drummen, Molecules, 2012, 17, 14067–14090 CrossRef CAS PubMed.
- S. P. Sau and P. J. Hrdlicka, J. Org. Chem., 2012, 77, 5–16 CrossRef CAS PubMed.
- G. Bains, A. B. Patel and V. Narayanaswami, Molecules, 2011, 16, 7909–7935 CrossRef CAS PubMed.
- A. M. Breul, M. D. Hager and U. S. Schubert, Chem. Soc. Rev., 2013, 42, 5366–5407 RSC.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- M. E. Ostergaard and P. J. Hrdlicka, Chem. Soc. Rev., 2011, 40, 5771–5788 RSC.
- A. Okamoto, Y. Saito and I. Saito, J. Photochem. Photobiol., C, 2005, 6, 108–122 CrossRef CAS PubMed.
- J. Xie, M. Ménand, S. Maisonneuve and R. Métivier, J. Org. Chem., 2007, 72, 5980–5985 CrossRef CAS PubMed.
- L. Zophel, V. Enkelmann and K. Mullen, Org. Lett., 2013, 15, 804–807 CrossRef PubMed.
- J. M. Casas-Solvas, J. D. Howgego and A. P. Davis, Org. Biomol. Chem., 2014, 12, 212–232 CAS.
- C. X. Yao, H. B. Kraatz and R. P. Steer, Photochem. Photobiol. Sci., 2005, 4, 191–199 CAS.
- M. Ottonelli, M. Piccardo, D. Duce, S. Thea and G. Dellepiane, J. Phys. Chem. A, 2012, 116, 611–630 CrossRef CAS PubMed.
- Y. Niko, Y. Hiroshige, S. Kawauchi and G. I. Konishi, J. Org. Chem., 2012, 77, 3986–3996 CrossRef CAS PubMed.
- Y. Niko, Y. Hiroshige, S. Kawauchi and G. I. Konishi, Tetrahedron, 2012, 68, 6177–6185 CrossRef CAS PubMed.
- Y. Niko, Y. Cho, S. Kawauchi and G.-i. Konishi, RSC Adv., 2014, 4, 36480–36484 RSC.
- Y. Niko and G. I. Konishi, Macromolecules, 2012, 45, 2327–2337 CrossRef CAS.
- F. Seela and S. A. Ingale, J. Org. Chem., 2010, 75, 284–295 CrossRef CAS PubMed.
- D. C. Knapp, J. D'Onofrio and J. W. Engels, Bioconjugate Chem., 2010, 21, 1043–1055 CrossRef CAS PubMed.
- S. Kodate, H. Wada, I. Suzuka, A. Okamoto, K. Tainaka, Y. Saito and I. Saito, Nucleic Acids Symp. Ser., 2004, 2006, 211–212 Search PubMed.
- S. T. Gaballah, Y. H. A. Hussein, N. Anderson, T. Q. T. Lian and T. L. Netzel, J. Phys. Chem. A, 2005, 109, 10832–10845 CrossRef CAS PubMed.
- A. Okamoto, K. Kanatani and I. Saito, J. Am. Chem. Soc., 2004, 126, 4820–4827 CrossRef CAS PubMed.
- J. Hwang, M. G. Choi, S. Eor and S.-K. Chang, Inorg. Chem., 2012, 51, 1634–1639 CrossRef CAS PubMed.
- S. Eor, J. Hwang, M. G. Choi and S.-K. Chang, Org. Lett., 2011, 13, 370–373 CrossRef CAS PubMed.
- S. P. Anthony, ChemPlusChem, 2012, 77, 518–531 CrossRef CAS.
- M. Shimizu and T. Hiyama, Chem.–Asian J., 2010, 5, 1516–1531 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- D. Plazuk, J. Zakrzewski, A. Rybarczyk-Pirek and S. Domagała, J. Organomet. Chem., 2005, 690, 4302–4308 CrossRef CAS PubMed.
- A. Wesolowska, L. Gros, S. Westerlich and T. S. Jagodzinski, ARKIVOC, 2008, 239–255 CrossRef CAS.
- T. Jagodzinski, Org. Prep. Proced. Int., 1990, 22, 755–760 CrossRef CAS.
- E. P. Papadopoulos, J. Org. Chem., 1976, 41, 962–965 CrossRef CAS.
- A. Wrona-Piotrowicz, D. Plazuk, S. Domagala and J. Zakrzewski, ARKIVOC, 2012, 412–420 CrossRef CAS.
- D. Plazuk, J. Zakrzewski, A. Rybarczyk-Pirek and S. Domagala, J. Organomet. Chem., 2005, 690, 4302–4308 CrossRef CAS PubMed.
- S. Aki, T. Fujioka, M. Ishigami and J.-i. Minamikawa, Bioorg. Med. Chem. Lett., 2002, 12, 2317–2320 CrossRef CAS.
- A. Corsaro and V. Pistara, Tetrahedron, 1998, 54, 15027–15062 CrossRef CAS.
- I. Mohammadpoor-Baltork, M. M. Sadeghi and K. Esmayilpour, Phosphorus, Sulfur Silicon Relat. Elem., 2003, 178, 61–65 CrossRef CAS.
- I. Mohammadpoor-Baltork, M. M. Sadeghi and K. Esmayilpour, Synth. Commun., 2003, 33, 953–959 CrossRef CAS PubMed.
- N. S. S. Kumar, S. Varghese, C. H. Suresh, N. P. Rath and S. Das, J. Phys. Chem. C, 2009, 113, 11927–11935 CAS.
- M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963–4976 RSC.
- Z. Zhao, S. Chen, J. W. Y. Lam, Z. Wang, P. Lu, F. Mahtab, H. H. Y. Sung, I. D. Williams, Y. Ma, H. S. Kwok and B. Z. Tang, J. Mater. Chem., 2011, 21, 7210–7216 RSC.
- R. Katoh, K. Suzuki, A. Furube, M. Kotani and K. Tokumaru, J. Phys. Chem. C, 2009, 113, 2961–2965 CAS.
- T. Seko, K. Ogura, Y. Kawakami, H. Sugino, H. Toyotama and J. Tanaka, Chem. Phys. Lett., 1998, 291, 438–444 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1006140. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra07045c |
| This journal is © The Royal Society of Chemistry 2014 |