
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Salt-confinement enables production of nitrogen-doped porous carbons in an air oven†
Martina
Ambrogi
a,
Yongjun
Men
a,
Frank
Polzer
b and
Jiayin
Yuan
*a
aDepartment of Colloid Chemistry Max Planck Institute of Colloids and Interfaces Am Mühlenberg 1, D-14476 Potsdam, Germany. E-mail: jiayin.yuan@mpikg.mpg.de
bMaterials Science & Engineering University of Delaware, 217C Du Pont Hall, 19716 Newark, USA
First published on 26th August 2014
Abstract
The production of nitrogen-doped porous carbons in an air oven at 750 °C with high surface area (≥500 m2 g−1), carbonization yield up to 35 wt% and tuneable nitrogen content from ionic liquids was reported. It utilized inorganic halide salts as the reaction medium, the activation agents, and the physical barrier. This method was successfully expanded to recyclable sea salt and natural sources, such as nucleobase (adenine) and biomass (oak leaves).
1. Introduction
Nitrogen-doped porous carbons have attracted considerable interest in the past decade due to their unique physical and chemical properties and a wide application scope in the fields of catalysis, environment techniques and energy generation and storage.1,2 The doping of nitrogen atoms in the carbon framework is well known to adjust the physical properties of carbon materials by increasing the overall electron density at the Fermi level, which improves the electric conductivity and oxidation stability.3,4 In addition, nitrogen doping enhances the basicity of carbons and makes them highly attractive for catalytic applications, for example, methanol oxidation, anchoring of catalytically active metal nanoparticles, and oxygen reduction reactions, only to name a few.5–14 Meanwhile, the unusual benefit received via nitrogen doping in carbons can be further amplified in a porous configuration that bears a high surface area to accelerate the mass/energy transport and boost their materials performance in electrochemical devices, such as lithium-ion batteries, electrochemical double layer capacitors and fuel cells.2,14,15Two fundamental synthetic approaches to introduce nitrogen into carbons can be distinguished, the thermal treatment of carbon or its precursor under ammonia, or the straightforward carbonization of nitrogen-rich precursors.1,16–21 The former deposits nitrogen atoms preferentially on the carbon surface, while the latter provides incorporation of structural nitrogen atoms into the bulk. Independent of the choice of these two methods, production of nitrogen-doped porous carbon under an oxygen-poor atmosphere is considered as a prerequisite, since the presence of oxygen at the carbonization temperature (typically 600 °C or above) will heavily attack the carbon product and turn it into gaseous compounds, usually CO or CO2. This oxidation effect is much strengthened for porous carbons, where enormous surface area is available to undergo and accelerate the oxidation reaction. A literature survey indicates that the production of porous carbons with high specific surface area, SBET (≥500 m2 g−1) directly in air is a rare case, not to mention the possible doping of nitrogen. Stucky et al. reported recently that an ultrasonic spray pyrolysis method with consecutive thermal treatment of confined core–shell microparticles could create porous carbon microspheres (nitrogen-free). In this case the porous carbon core of micrometer size was confined and protected by a thin, dense TiO2 shell for the pyrolysis in air.22 However, the necessity of particular equipment, complex design of the core–shell morphology, and the post-synthesis treatment to etch the TiO2 scaffold limits the advantage of this process over the common preparation methods under inert atmosphere.23 In fact not only the noble gas as protecting atmosphere but also the engineering setups increase the final production cost and complexity. From this point of view, production of nitrogen-doped porous carbons bearing a high SBET without noble gas protection is not only scientifically challenging but also of practical values.
In this work a sustainable and easy-to-operate method to produce porous nitrogen-doped carbon materials in air via a salt-assisted approach is demonstrated. Specifically speaking, common inorganic halide salts, NaCl and ZnCl2 were employed as the high-temperature ionic solvent, the activation agents to introduce pores into carbons, and the protecting barrier to confine a local oxygen-poor environment. Very interestingly this concept can be straightforward extended to natural sources, such as sea salt as reaction medium, and nucleobase or oak leaves as carbon precursor. The general synthetic route is illustrated in Fig. 1.
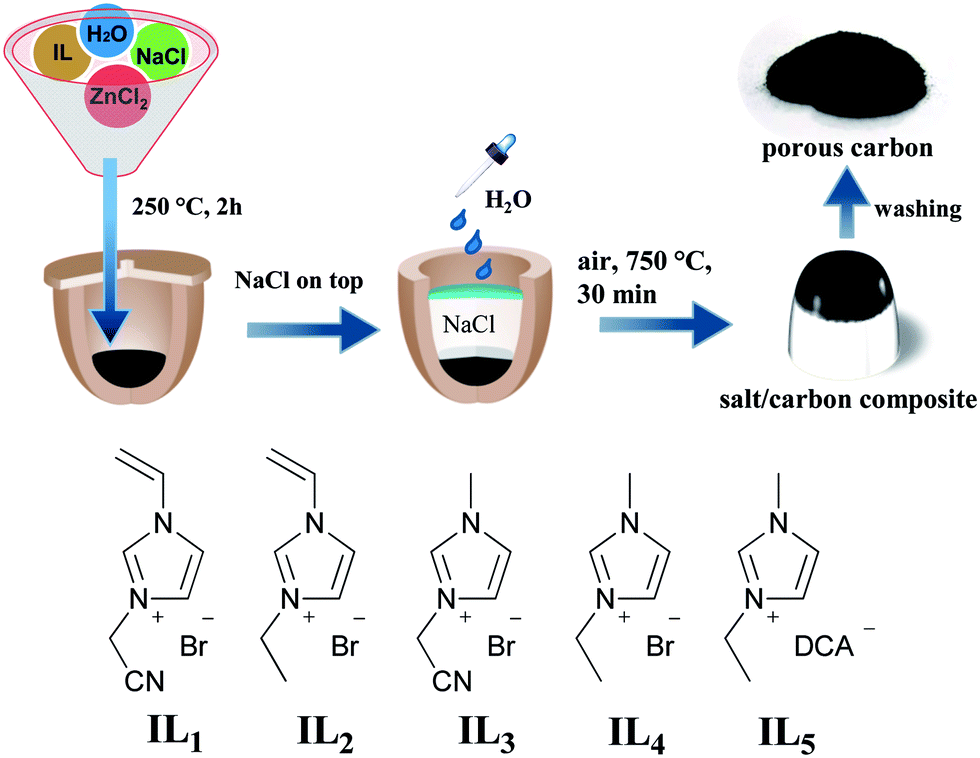 | ||
| Fig. 1 Schematic representation of the carbonization process in an air oven. The chemical structures of the used ILs are illustrated beneath. DCA represents the dicyanamide anion. | ||
2. Experimental
2.1 Materials
All chemicals were used as supplied. Reagents for the syntheses of IL4 and IL5 were purchased from IoLiTec company, and the rest were synthesized by ourselves according to our previous route.24 The salts NaCl (99%) and ZnCl2 (99%) were purchased from Sigma-Aldrich. All solvents used were of analytic grade. Ultrapure Milli-Q water was used in all experiments. Oak leaves were collected from a thicket located in Potsdam, Germany.2.2 Synthesis of nitrogen-doped porous carbon from ILs
In a general procedure 1.5 g of carbon precursors was mixed with salt combination of NaCl and ZnCl2 or Red Sea salt in a defined weight ratio listed in Table 1. The starting mixture was dissolved in 6 ml of water in a porcelain crucible, heating at 60 °C for 15 minutes under stirring. The sample was heated up to 250 °C (heating rate 4 °C min−1) in air oven and the temperature was maintained stable for 2 hours. Afterwards the sample was directly covered with NaCl or sea salt (roughly 60 g). 3 ml of water were dropped on the surface in order to melt a thin layer of salt and then let it evaporate at room temperature for 30 minutes. The sample was placed in an air oven previously heated at 750 °C for the desired time and after that the sample was pulled out of the oven and cooled down to room temperature. The salt was then simply dissolved in water and the resulting carbon was treated with 1 N HCl aqueous solution for two days in order to remove final impurities from the pores. The product was separated by centrifugation and washed several times with water until neutral pH and then with mixtures of water–ethanol. The as-prepared carbon material was finally dried under vacuum at 80 °C overnight.| Entry | Sample | S BET (m2 g−1) | Pore volume (cm3 g−1) | Yield (wt%) | C (wt%) | N (wt%) | H (wt%) | |||
|---|---|---|---|---|---|---|---|---|---|---|
a SS: sea salt. These are the 3 recycling tests. IL1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SS weight ratio is 1:1.
b Using adenine and oak leaves (OL) as the carbon precursor. :SS weight ratio is 1:1.
b Using adenine and oak leaves (OL) as the carbon precursor.
|
||||||||||
| 1 | IL1Z0S1T30 | 207 | 0.094 | 20.6 | 74.2 | 13.4 | 1.9 | |||
| 2 | IL1Z0.1S1T30 | 412 | 0.172 | 31.2 | 68.0 | 13.5 | 2.3 | |||
| 3 | IL1Z0.3S1T30 | 950 | 0.453 | 29.4 | 68.5 | 15.2 | 2.5 | |||
| 4 | IL1Z0.5S1T30 | 811 | 0.690 | 28 | 71.1 | 13.6 | 2.4 | |||
| 5 | IL1Z0.75S1T30 | 886 | 0.695 | 19.8 | 67.3 | 13.9 | 2.9 | |||
| 6 | IL1Z1 S1T30 | 894 | 0.588 | 8.4 | 67.7 | 12.0 | 2.6 | |||
| 7 | IL1Z0.3S0T30 | 419 | 0.186 | 34.3 | 64.2 | 14.9 | 2.5 | |||
| 8 | IL1Z0.3S0.3T30 | 809 | 0.391 | 31.5 | 67.2 | 15.4 | 2.6 | |||
| 9 | IL1Z0.3S0.6T30 | 747 | 0.538 | 31.9 | 68.2 | 15.4 | 2.7 | |||
| 10 | IL1Z0.3S2T30 | 805 | 0.397 | 33.1 | 67.1 | 16.0 | 2.9 | |||
| 11 | IL1Z0.3S1T15 | 812 | 0.477 | 28.3 | 64.5 | 15.3 | 2.7 | |||
| 12 | IL1Z0.3S1T60 | 990 | 0.569 | 19.3 | 68.2 | 12.3 | 2.6 | |||
| 13 | IL1Z0.3S1T120 | 1130 | 0.692 | 8.4 | 70.6 | 10.6 | 2.4 | |||
| 14 | IL2Z0.3S1T30 | 502 | 0.273 | 11.5 | 67.6 | 12.7 | 2.3 | |||
| 15 | IL3Z0.3S1T30 | 920 | 0.543 | 20.5 | 60.8 | 21.8 | 2.7 | |||
| 16 | IL4Z0.3S1T30 | n.d. | n.d. | <1 | n.d. | n.d. | n.d. | |||
| 17 | IL5Z0.3S1T30 | 562 | 0.383 | 4.9 | 43.6 | 32.1 | 3.1 | |||
| 18a | IL1-SS-T30_1° | 750 | 0.406 | 10.0 | 57.7 | 10.9 | 1.98 | |||
| 19a | IL1-SS-T30_2° | 560 | 0.224 | 13.6 | 55.2 | 12.6 | 1.80 | |||
| 20a | IL1-SS-T30_3° | 694 | 0.255 | 13.6 | 62.9 | 12.5 | 2.00 | |||
| 21b | AdenineZ0.3S1T30 | 412 | 0.172 | 32.0 | 42.6 | 31.0 | 2.47 | |||
| 22b | OL-SS-T30 | 600 | 0.453 | 23.8 | 61.3 | 1.64 | 1.16 | |||
2.3 Synthesis of nitrogen-doped porous carbon from IL1 under N2 flow, with and without ceiling
The mixture of IL1:ZnCl2:NaCl (weight ratio of 1:0.3:1) in water was dried in air oven at 250 °C for 2 hours, and the resulting carbonaceous matrix was then covered with fined grinded NaCl. The starting material was placed in the oven at room temperature and flushed with nitrogen flow for one hour before starting the heating in order to avoid eventually oxidation reactions during thermal treatment. The temperature was then increased with a heating rate of 10 °C min−1 up to 750 °C and the sample was held at this temperature for 30 minutes. After the treatment the sample was left inside the chamber until the temperature was cooled down naturally. The same purification procedure as the carbon produced in air oven was performed (see Section 2.2). When carbonization under nitrogen without NaCl ceiling on the top was performed, the procedure is exactly the same, except the sample was directly carbonized after the drying process (heating rate 10 °C min−1, holding time 30 minutes).
2.4 Synthesis of nitrogen-doped porous carbon from adenine
We used the same procedure reported in Section 2.2 with adenine instead of IL. The weight ratio is adenine:ZnCl2:NaCl = 1:0.3:1.
2.5 Synthesis of porous carbon from oak leaves and red sea salt
Oak leaves were firstly washed with water and then dried at 80 °C overnight. After grinded 1.5 g of leaves, a 1:1 weight ratio of sea salt was added and mixed carefully. The solid mixture was transferred into a crucible and fresh sea salt (roughly 60 g) was used to generate the confinement. 3 ml of water were dropped on the surface to melt a thin layer of salt and then it was dried at room temperature for 30 minutes. The sample was then placed directly at 750 °C in air oven for 30 minutes. The work-up of the product purification was the same as reported for ILs (see Section 2.2).
2.6 Characterization methods
Nitrogen sorption measurements were performed on Quantachrome Autosorb-1 at 77 K (liquid nitrogen temperature). Samples were degassed prior to the measurements at 150 °C for 20 hours. The specific surface area is calculated according to the Brunauer–Emmett–Teller (BET) equation, while quenched solid density functional theory (QSDFT) for cylindrical pores is applied for the evaluation of pore size distribution. This latter method takes in consideration the surface roughness and heterogeneity of the material, representing a more precise and realistic model for the surface area interpretation here. Combustion elemental analysis (C/H/N content) was carried out for all samples using Vario Micro setup (version 1.4.1). Thermal gravimetric analyses (TGA) were performed on a Netzsch TG209-F1 apparatus at a heating rate of 10 °C min−1. High resolution transmission electron microscopy (HR-TEM) and elemental mapping investigation were carried out on a JEOL JEM-2200FS instrument (JEOL GmbH, Eching, Germany). For sample preparation, one drop of the sample dispersion in ethanol was placed on a 200 mesh carbon coated copper grid and dried in air. TEM investigations were carried out at temperature around 90 K, operating at an acceleration voltage of 200 kV. All images were recorded digitally by a bottom-mounted 4k*4k CMOS camera system using low dose conditions (TemCam-F416, TVIPS, Gauting, Germany) and processed with a digital imaging processing system (EM-Menu 4.0, TVIPS, Gauting, Germany). X-ray diffraction experiments were done with a Bruker D8 diffractometer using Cu Kα radiation (λ = 0.154 nm) and a scintillation counter. XPS data were obtained on Thermo ESCALAB250 instrument with a monochromatized Alkaline source (200 W). A Controller P320 professional furnace (Nabertherm Company) was used to perform all the carbonization processes. Porcelain crucibles of 40 mm in diameter, purchased from VWR Company, were used for all the syntheses. FT-IR spectra were recorded on a Varian1000 FT-IR spectrometer. SEM image was obtained on a LEO 1550-Gemini instrument with platinum sputtering.3. Results and discussion
Imidazolium ionic liquids (ILs) are employed as model carbon precursors in this preparation process because of their negligible vapor pressure, high thermostability and the significant presence of nitrogen in the final carbon.6 Some recent reports have proven that a carbonization yield of 10–30 wt% at 800–1000 °C and a tunable nitrogen content in terms of the chemical design can be reached via an IL route.6,9 Particularly from a structure point of view, the network-forming groups in ILs are known to build up crosslinked intermediates to improve the structural stability and correspondingly the carbonization yield. In this study ILs with different functional groups, shown in Fig. 1, are tested. In a typical run a mixture of IL, NaCl and ZnCl2 in a defined weight ratio is homogeneously dissolved in water in a porcelain crucible, and then thermally treated at 250 °C in air for 2 h. This step dried the mixture and produced a solid black solution. In the presence of cyano and vinyl groups in ILs, trimerisation and the thermal polymerization occurred in parallel, which formed a crosslinked network (Fig. S1†) and favorably stabilized the carbon intermediate. Afterwards, finely grinded NaCl powder was packed directly above the black composite in the crucible. It is important to spread a few water drops to partially wet the upper NaCl surface, which becomes highly dense after thermal evaporation of water to reach a maximum space confinement. The NaCl ceiling acts here as a solid physical barrier to minimize the oxygen entrance. In addition, this confinement can retard the diffusion of the gasified decomposition products, mostly the hydrocarbons, out of the reactor, thus raising the overall carbonization yield.The carbonization took place via inserting the crucible reaction in an air oven at 750 °C, i.e. below the melting point of NaCl (801 °C) for 30 min. At this stage, several events occur parallel. ZnCl2 (m.p. ∼292 °C) melts and dissolves NaCl at the bottom of the crucible, creating an ionic, homogeneous, liquid environment for the carbon formation, in which ZnCl2 initiates the carbon activation reaction. It should be clarified that although the NaCl top layer remains solid at the carbonization temperature to create a closed environment, the bottom part may join the molten salt due to the direct contact with the ZnCl2/NaCl liquid mixture. Finally, the mixture was pulled out of the oven for cooling down to room temperature. The dark mixture was treated by acidic water before completely dried.
Table 1 collects characterization data of the as-synthesized carbon products. To reach a comprehensive view of the scope and applicability of this method, we systematically varied the experimental parameters, such as the amount/ratio of ZnCl2 and NaCl, the carbonization time and the IL chemical structure. The SBET, pore volume, carbonization yield and elemental analysis results of the final carbon product were measured and recorded. For the sake of conciseness and clearness, the carbon products derived from ILs were denoted as ILwZxSyTz, in which w, x, y and z stand for the IL type, the wt% of ZnCl2 with regard to IL, the wt% of NaCl, and the carbonization time at 750 °C in min, respectively. As shown in Table 1, porous carbons with large SBET and high nitrogen doping were indeed received via this route. As a representative product, IL1Z0.30S1T30 (Entry 3 in Table 1), which used IL1 as the carbon precursor and a salt mixture of 30 wt% of ZnCl2 and 100 wt% of NaCl, was chosen to demonstrate the structural properties of the as-synthesized porous carbons. Fig. 2A displayed the nitrogen sorption isotherm measured at 77 K. A type-I isotherm is clearly identified and proves the dominant existence of micropores and a low fraction (<10%) of small mesopores in this carbon product. The pore volume is 0.453 cm3 g−1, and a SBET of 950 m2 g−1 is reached. Quenched solid density functional theory (QSDFT) is used to determine the average pore size, which is in the range of 1–5 nm (Fig. S5†, red curve). It should be mentioned that in some carbon products prepared at different conditions listed in Table 1, their sorption isotherms (Fig. S5–S8†) have a broad hysteresis at high values of relative pressure. This indicates interstitial pore formation that is related to the morphology of the carbon particle aggregation discussed later. The crystalline status is characterized via X-ray diffraction (XRD) measurements. As shown in Fig. 2B, characteristic bands at 26°, 43° and 81° were observed, which are assigned to the (002), (100) and (110) diffraction bands of graphitic phase, respectively. The weak intensity at 81° implies that the obtained carbon lacks of a long range order, i.e. turbostratic.
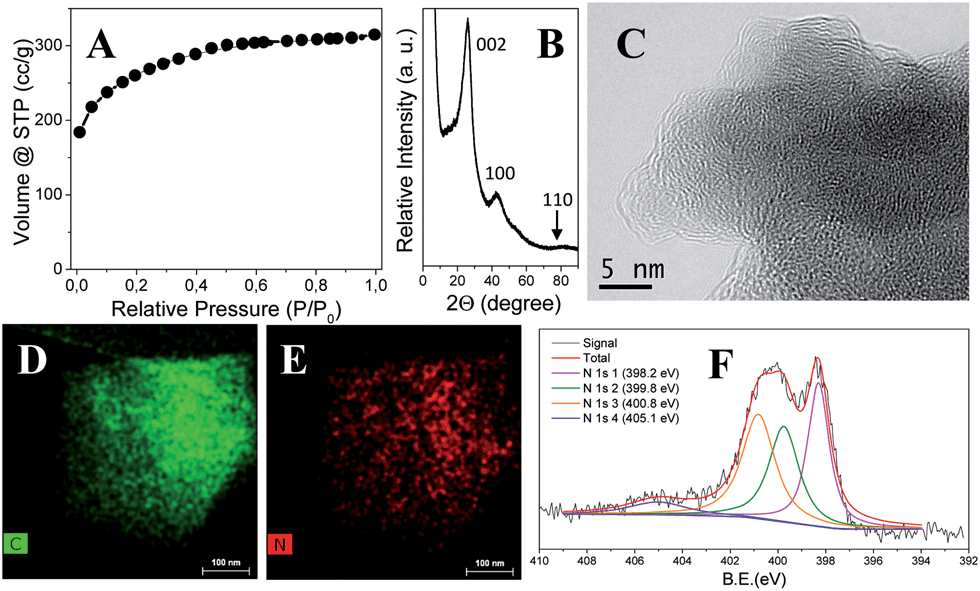 | ||
| Fig. 2 Characterization of the carbon product IL1Z0.30S1T30: (A) nitrogen adsorption/desorption isotherm curve; (B) XRD pattern; (C) HRTEM image; (D and E) elemental mapping of carbon and nitrogen elements spectroscopy, respectively (obtained via SEM-EDX spectroscopy); (F) XPS spectrum of the nitrogen element. | ||
To visualize the local atomic order, high resolution transmission electron microscopy (HR-TEM) was employed. Irregularly bent planes of various lengths are clearly visible in Fig. 2C. Scanning and transmission electron microscopy shows that IL1Z0.30S1T30 is composed of aggregated primary carbon nanoparticles of 20–50 nm in size (Fig. S2†). This kind of morphology of carbon particles was also observed in many other carbons in Table 1. The packing of the carbon nanoparticles densely together creates interstitial voids, responsible for the broad hysteresis at high values of relative pressure of the nitrogen sorption isotherms of some carbon products. Combustion elemental analysis reveals a high nitrogen content of 15 wt%, which is homogeneously distributed, as confirmed by elemental mapping of the C and N atoms based on energy dispersive X-ray spectroscopy (EDX, Fig. 2D and E). The elemental composition of the sample surface was further checked by X-ray photoelectron spectroscopy (XPS) which depicts the presence of carbon, nitrogen and oxygen in 77.4, 14.1 and 8.5 wt%, respectively (Fig. S3†). The C and N values are close to that obtained by combustion elemental analysis method, which agrees with the EDX analysis on a homogenous distribution of the elements across the sample. A close view of the chemical states of N1s is reported in Fig. 2F. Typical binding energies of pyridinic (30.6%, 398.2 eV), pyrrolic (28.1%, 399.8 eV), quaternary – graphitic (34.0%, 400.8 eV) and oxidic-N species (7.3%, 405.1 eV) are found, proving the presence of structural nitrogen atoms intercalated in the material matrix. The existence of an oxidic-N species is a nature outcome of the carbonization process, where a small fraction of oxygen is incorporated (Fig. S4†). For the sake of completeness, control experiments under nitrogen flow were also performed (named “N2 carbon”). In detail, the production of the sample IL1Z0.30S1T30 was tested under inert gas flow both with and without NaCl ceiling. The resultant N2 carbons showed lower surface areas and pore volumes compared with the carbon obtained in air oven. Specifically 766 m2 g−1 and 0.371 cm3 g−1 for the salt-confined N2 carbon and 763 m2 g−1 and 0.328 cm3 g−1 for the N2 carbon without NaCl ceiling were obtained (Fig. S11 and S12,† respectively). The nitrogen content of N2 carbons was similar to that produced in air oven (15.7 wt% for the salt-confined N2 carbon and 14.7 wt% for the N2 carbon without NaCl ceiling), while a slightly higher yield was observed (34.5 wt% for the salt-confined N2 carbon and 36.4 wt% for the N2 carbon without NaCl ceiling).
As a commonly used activation agent, the effect of ZnCl2 is first investigated (Entry 1–6 in Table 1, Fig. 3A and S5†). While fixing the weight ratio of IL1:NaCl to 1:1 and varying that of ZnCl2:IL1 gradually from 0 to 100 wt%, the SBET increased dramatically upon the initial addition of ZnCl2 until 30 wt%. Beyond this value, it remains constant around 900 m2 g−1. In comparison, the ZnCl2-effect on the carbonization yield is more complex. Addition of a small portion (≤10 wt%) leads to a high carbonization yield up to 31 wt%. Above 10 wt% of ZnCl2, the carbonization yield drops. At the maximum ZnCl2 addition (100 wt%) in the tests (Entry 6 in Table 1), an unfavorable low yield of only 8.4 wt% was received. The complex effect of ZnCl2 is caused by the double role of ZnCl2 in the carbonization process. It serves as the activation agent for the pore formation, which creates surface area and suppresses the yield. Meanwhile, it catalyzes the trimerisation of the cyano groups, which construct stable intermediates to reduce the structural fragmentation at the carbonization temperature, thus increasing the yield. Our data supports that a low fraction of ZnCl2 promotes the formation of crosslinked networks, and overcompensates the weight loss caused by the activation. Too much addition of ZnCl2 however makes the etching process more dominant. It is not surprising that the highest yield is found at a balanced content of ZnCl2, here 10 wt% (Entry 2 in Table 1).
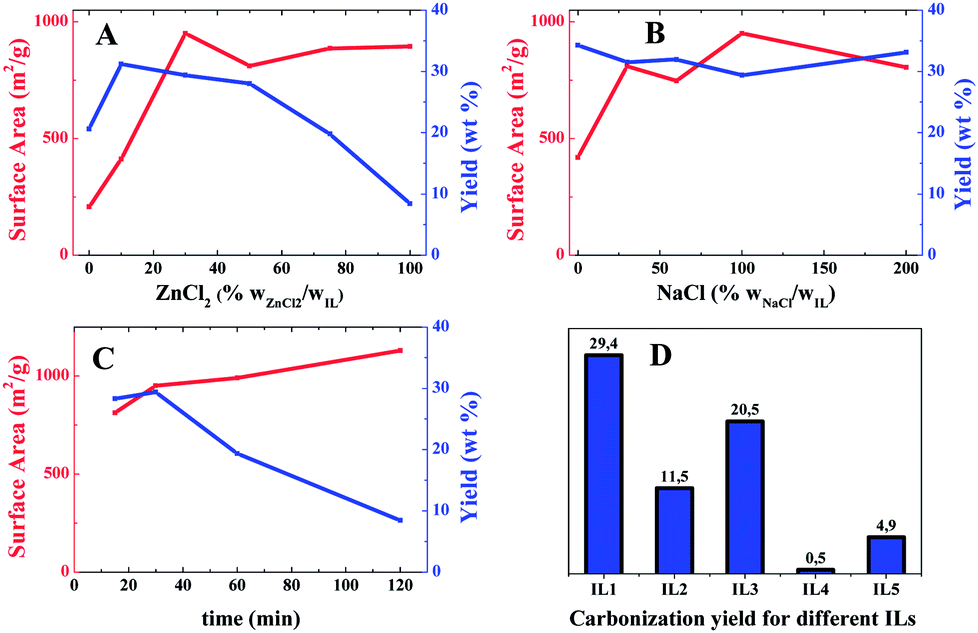 | ||
| Fig. 3 The variation of the specific surface area and the carbonization yield of the nitrogen-doped porous carbons prepared at different concentrations of ZnCl2 (A), NaCl (B) and different period (C). Yields from different IL precursors are also reported (D). | ||
It is curious to find that even in the absence of ZnCl2 (Entry 1 in Table 1), a porous carbon with a medium SBET of 200 m2 g−1 was obtained. This finding tells that in the current process, NaCl seems to be not only a salt merely to dilute the ZnCl2 concentration, but also a co-activation agent, though weak. The role of NaCl (not the top NaCl layer) was thus thoroughly investigated (Entry 7–10 in Table 1, Fig. S6†). As illustrated in Fig. 3B, in the presence of 30 wt% of ZnCl2, NaCl in a low fraction (30 wt%) increases the SBET from 419 up to 809 m2 g−1, indicating its participation in creation of porosity. Unlike ZnCl2, at even higher concentrations of NaCl above 30 wt%, the pore features and the yield of the final carbon products are unaffected.
As it is true for all carbonization processes, the dwelling time is crucial, necessary means to control the physical properties of the carbons. To understand the pore development, experiments with several duration times were performed. We fixed the IL1:ZnCl2:NaCl ratio to 1:0.3:1 (Entry 11–13 in Table 1, Fig. 3C and S7†). It was found that the SBET increases from 813 (15 min) to 1130 m2 g−1 (2 h), indicating the activation continuously introduces more porosity. In contrast, the carbonization yield decreases from 28.3 wt% to 8.4 wt% gradually due to the prolonged etching time and a possible entrance of oxygen into the reactor upon prolonged dwelling times. It is particularly worth noting that only after 15 min the SBET jumps already to 812 m2 g−1, implying a fast conversion of the precursor to porous carbons.
The chemical structure of ILs is important to modulate the physical property of the carbon product. Besides IL1, four other imidazolium-based ILs (Fig. 1) with different functional groups are tested in order to reveal the relationship between the IL chemical structure and the physical feature of the as-synthesized carbons. A carbonization time of 30 min, and a starting mixture of IL:NaCl:ZnCl2 in ratio of 1:1:0.3 were fixed for all runs (Entries 14–17 in Table 1, Fig. 3D and S8†).
When the cyano group in IL1 is replaced by a methyl group in IL2, a yield of 11.5 wt%, and a surface area of 502 m2 g−1 was observed (Entry 14 in Table 1). Both values are significantly lower than that of IL1. It proves the necessity to bring the cyano group into the IL chemical structure. Keeping the cyano functionality unchanged but substituting the vinyl with a methyl group, i.e. IL3, a comparable surface area of 920 m2 g−1 is obtained (Entry 15). Yet the carbonization yield drops by 9% to 20.5 wt%. As discussed above, the absence of the vinyl group fails to generate polymer chains that undergo crosslinking by the cyano groups, thus amplifying the structural fracture effect. A beneficial of IL3 is the high nitrogen content, 22 wt%. When both cyano and vinyl groups are absent, i.e. IL4, it is not astonishing to observe a carbonization yield below 1 wt% (Entry 16), a useless precursor for carbon production. Finally IL5, bearing a cyano-containing anion was tested. A significant high nitrogen content, 32 wt% (C/N ratio of 1.4) was observed. Unfortunately, the yield is as low as 4.9 wt%. Therefore, both the vinyl and cyano groups on the cation are of key importance.
The central spirit of carrying out carbonization process without noble gas protection is to create a local oxygen-poor milieu to run oxygen-sensitive high temperature reaction. We should note that an absolute oxygen-free circumstance throughout the entire carbonization process is only idealistic. Though the top NaCl floor is dense, microchannels do exist. The reaction mixtures after 10 and 30 min were illustrated in Fig. 4. The 10 min sample displays, besides a black IL/ZnCl2/NaCl bottom body, a grey top NaCl zone. This is an indication that following the initial violent decomposition of the ILs at 750 °C, the gaseous fracture compounds, majorly the carbohydrates, build up inner pressure to force the gas diffusion through the interstice in the top NaCl ceiling. The grey color thus stems from the carbon deposition of the gaseous compounds. This overpressure blocks the intrusion of the oxygen into the reactor and guarantees a true inert atmosphere at this stage. The white color of the top NaCl zone after 30 min however implies that after the initial conversion of the ILs quickly into carbons, the major reaction switches to the chemical activation by ZnCl2, a relatively mild process. The high pressure built up at the initial IL thermal decomposition stage vanished, which enables the (partial) entrance of oxygen, though slow, through the microchannels in the NaCl zone into the reactor and oxidizes the formed carbons into CO or CO2. This is in part responsible for the low yield upon elongated carbonization (Fig. 3C). From this point of view, 30 min is an ideal reaction period to minimize the oxygen disturbance. It should be noted that due to the oxygen penetration, it may very possibly create a gradient oxidative carbon product along the sample from the top to the bottom.
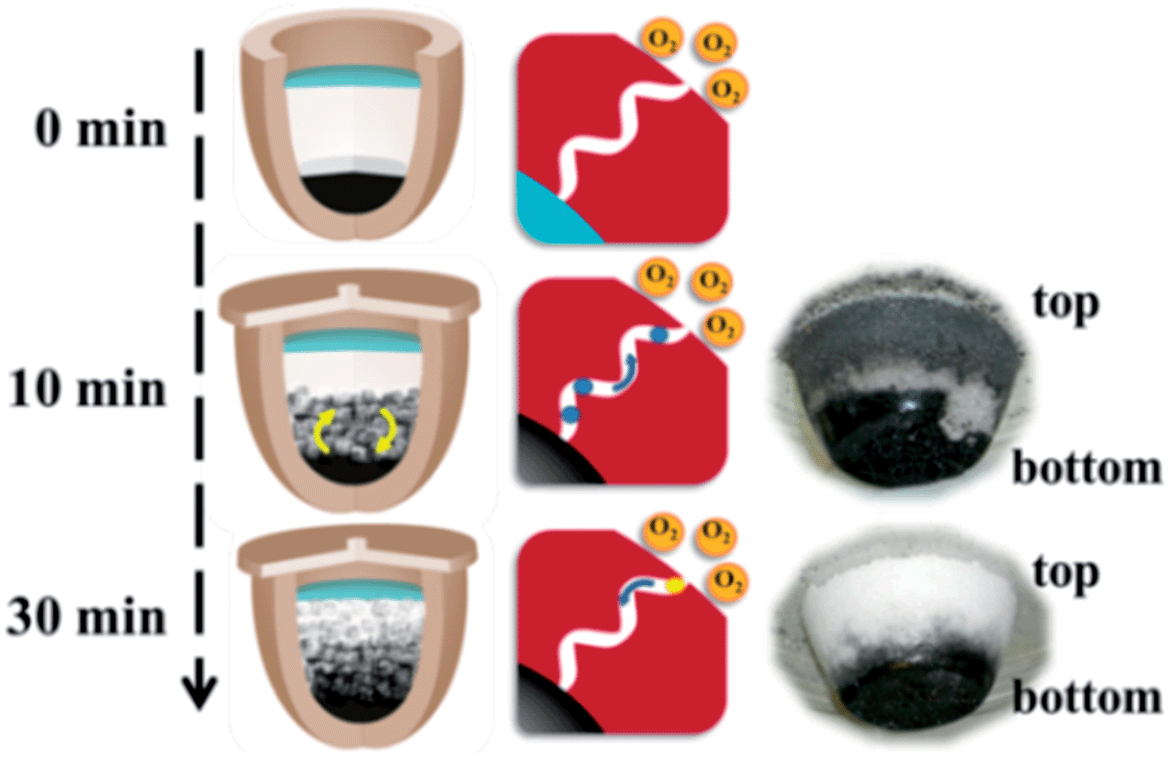 | ||
| Fig. 4 Illustration of the reaction mixture after 10 and 30 min. The two photographs on the right side show the reaction mixture at 10 and 30 min. | ||
The implementation of the carbonization in air without noble gas protection relies on the isolation of the reactor from the surrounding by a salt shielding below its melting point. We were motivated to modify this concept into a general and more sustainable methodology. Two key factors attract our attention, the salt mixture and the carbon precursor. In the former case, we could use sea salt to replace ZnCl2/NaCl. The sea salt we tested is provided from RedSea© and it is usually used to replicate reef water system. Since the dominant fraction is NaCl, it serves favorably as the ceiling layer. Meanwhile, due to the coexistence of a small fraction of MgCl2 (3.3 wt%), CaCl2 (1.1 wt%) and KCl (1.0 wt%) species, it can conduct activation like ZnCl2. In the test run using IL1 as a carbon precursor, a yield of 10 wt% in 30 min and 11 wt% of nitrogen doping were obtained (Entry 18 in Table 1). The SBET is 750 m2 g−1, with pore volume of 0.406 cm3 g−1. These exciting results prove that the natural sea salt can replace the NaCl/ZnCl2 binary system. An additional merit of sea salt is the easy recyclability due to the identical salt composition all over the reactor. In a 3-recycle test, the sea salt preserves well its shielding and activation function (Entry 19 and 20 in Table 1 and Fig. S9†).
In the latter case to replace ILs as carbon precursor, we were motivated to employ natural raw materials. Nucleobases such as adenine, possess a high atomic N/C ratio, high oxidation stability, and suitable atomic N–C–N bonding motifs. They can be isolated from nature in larger quantities, and have been explored for functional carbon synthesis very recently.1 Here adenine was tested in the ZnCl2/NaCl system. The formed nitrogen-doped porous carbons showed a high carbonization yield of 32 wt%, a high nitrogen content of 31 wt% and a satisfactory SBET of 412 m2 g−1 (Entry 21 in Table 1, Fig. S10†). In addition to nucleobase, biomass is also applied. For example, the oak leaves have been successfully converted to porous carbon using sea salt (Entry 22 in Table 1, and Fig. S13†). As shown in Fig. 5, the oak leaves were collected and grinded with the sea salt into fine powders to reach a homogeneous mixture. Upon the carbonization procedure mentioned above (see Section 2.5), fine carbon powders with SBET of 600 m2 g−1 were achieved. The nitrogen sorption isotherm is shown in Fig. 5D. Beside a significant amount of micropores, the sample contains a considerable fraction of mesopores. The pore volume is 0.45 cm3 g−1, which contains a mesopore volume of 0.33 cm3 g−1. Its nitrogen content is relatively low, only 1.6 wt%, which comes from the proteins existing in the oak leaves (elemental analysis of fresh oak leaves showed nitrogen content of 1.1 wt%).
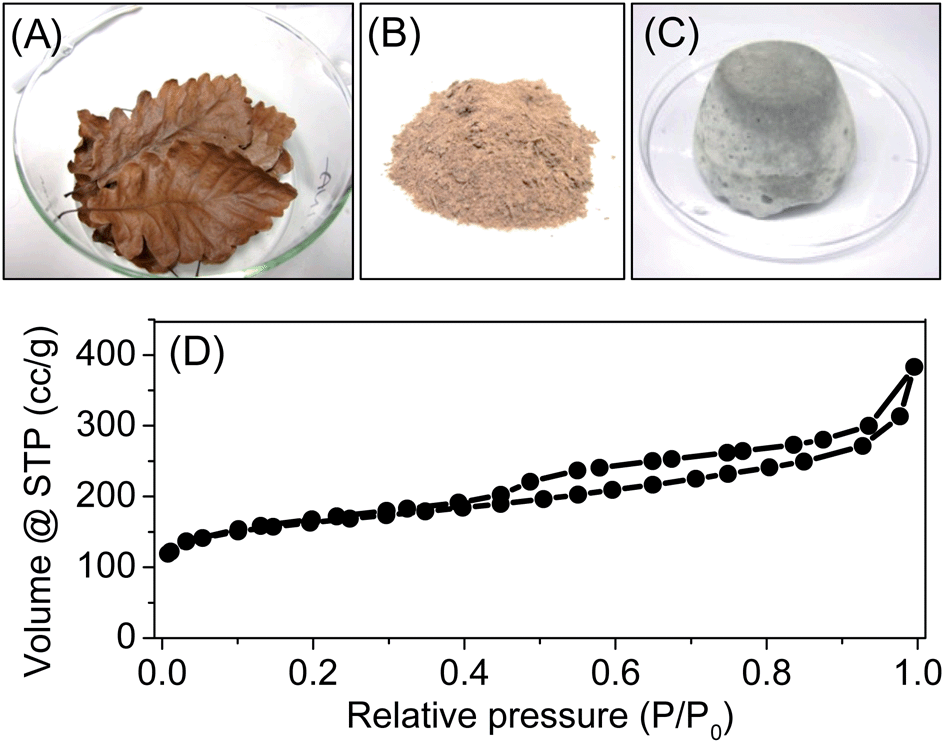 | ||
| Fig. 5 (A–C) Photographs of pristine oak leaves, grinded powder of oak leaves and sea salt, and the carbonization mixture after 30 min at 750 °C. (D) N2 sorption isotherm of the carbon prepared from the oak leaves. | ||
4. Conclusions
In summary, we introduced a general synthetic route to produce nitrogen-doped porous carbons with high surface area and tunable nitrogen content in an air oven. This conceptually straightforward production procedure is easy to implement. Not only the salt can be replaced by recyclable sea salt, but also the carbon precursor is accessible from natural sources, thus pointing out a more sustainable way for the nitrogen-doped carbon production. Furthermore, the method is expected to be able to conduct many other high temperature chemical processes in air instead of under inert gas environment, for instance, the preparation of metal nanoparticles, and the synthesis of metal carbide or nitride.Acknowledgements
F.P. thanked the CRC 951 of the German research Council and the Joint Lab for Structural Research within IRIS Adlershof for funding. J.Y. thanked Prof. M. Antonietti and Dr N. Fechler for helpful discussion. This work is financialized by the Max Planck Society and the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007–2013 under REA grant no. 289347.Notes and references
- W. Yang, T.-P. Fellinger and M. Antonietti, J. Am. Chem. Soc., 2010, 133, 206–209 CrossRef PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- D. P. Kim, C. L. Lin, T. Mihalisin, P. Heiney and M. M. Labes, Chem. Mater., 1991, 3, 686–692 CrossRef CAS.
- Y. Men, M. Siebenburger, X. Qiu, M. Antonietti and J. Yuan, J Mater. Chem. A, 2013, 1, 11887–11893 CAS.
- J. P. Paraknowitsch, A. Thomas and M. Antonietti, J. Mater. Chem., 2010, 20, 6746 RSC.
- J. S. Lee, X. Wang, H. Luo, G. A. Baker and S. Dai, J. Am. Chem. Soc., 2009, 131, 4596–4597 CrossRef CAS PubMed.
- Z.-S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- W. Li, Z. Zhang, B. Kong, S. Feng, J. Wang, L. Wang, J. Yang, F. Zhang, P. Wu and D. Zhao, Angew. Chem., Int. Ed., 2013, 52, 8151–8155 CrossRef CAS PubMed.
- J. P. Paraknowitsch, J. Zhang, D. Su, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 87–92 CrossRef CAS PubMed.
- S. Soll, T.-P. Fellinger, X. Wang, Q. Zhao, M. Antonietti and J. Yuan, Small, 2013, 9, 4135 CrossRef CAS PubMed.
- D. Kuzmicz, S. Prescher, F. Polzer, S. Soll, C. Seitz, M. Antonietti and J. Yuan, Angew. Chem., Int. Ed., 2014, 53, 1062–1066 CrossRef CAS PubMed.
- P. Zhang, J. Yuan, T.-P. Fellinger, M. Antonietti, H. Li and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 6028–6032 CrossRef CAS PubMed.
- Q. Zhao, T.-P. Fellinger, M. Antonietti and J. Yuan, J. Mater. Chem. A, 2013, 1, 5113–5120 CAS.
- J. Balach, H. Wu, F. Polzer, H. Kirmse, Q. Zhao, Z. Wei and J. Yuan, RSC Adv., 2013, 3, 7979–7986 RSC.
- K. K. R. Datta, V. V. Balasubramanian, K. Ariga, T. Mori and A. Vinu, Chem.–Eur. J., 2011, 17, 3390–3397 CrossRef CAS PubMed.
- W. Luo, B. Wang, C. G. Heron, M. J. Allen, J. Morre, C. S. Maier, W. F. Stickle and X. Ji, Nano Lett., 2014, 14, 2225–2229 CrossRef CAS PubMed.
- D. Kuzmicz, P. Coupillaud, Y. Men, J. Vignolle, G. Vendraminetto, M. Ambrogi, D. Taton and J. Yuan, Polymer, 2014, 55, 3423–3430 CrossRef CAS PubMed.
- M.-R. Gao, X. Cao, Q. Gao, Y.-F. Xu, Y.-R. Zheng, J. Jiang and S.-H. Yu, ACS Nano, 2014, 8, 3970–3978 CrossRef CAS PubMed.
- P. Chen, T.-Y. Xiao, Y.-H. Qian, S.-S. Li and S.-H. Yu, Adv. Mater., 2013, 25, 3192–3196 CrossRef CAS PubMed.
- G.-P. Hao, W.-C. Li, D. Qian and A.-H. Lu, Adv. Mater., 2010, 22, 853–857 CrossRef CAS PubMed.
- D. Wu, Z. Li, M. Zhong, T. Kowalewski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2014, 53, 3957–3960 CrossRef CAS PubMed.
- W. H. Suh, J. K. Kang, Y.-H. Suh, M. Tirrell, K. S. Suslick and G. D. Stucky, Adv. Mater., 2011, 23, 2332–2338 CrossRef CAS PubMed.
- C. Galeano, J. C. Meier, V. Peinecke, H. Bongard, I. Katsounaros, A. A. Topalov, A. Lu, K. J. J. Mayrhofer and F. Schüth, J. Am. Chem. Soc., 2012, 134, 20457–20465 CrossRef CAS PubMed.
- J. Yuan, C. Giordano and M. Antonietti, Chem. Mater., 2010, 22, 5003–5012 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra06886f |
| This journal is © The Royal Society of Chemistry 2014 |