DOI:
10.1039/C4RA05047A
(Paper)
RSC Adv., 2014,
4, 37705-37713
Immobilization of iron hydroxide/oxide on reduced graphene oxide: peroxidase-like activity and selective detection of sulfide ions†
Received
28th May 2014
, Accepted 12th August 2014
First published on 12th August 2014
Abstract
We prepared nanocomposites of amorphous iron hydroxide/oxide immobilized on reduced graphene oxide (FeOxH–rGO) with peroxidase-like activity for the detection of sulfide (S2−) ions. FeOxH–rGO nanocomposites were prepared by reaction of GO (size ∼ 300 nm) partially reduced by ultraviolet irradiation with Fe2+ in Tris–borate solution (5.0 mM, pH 7.0). The amorphous FeO(OH) and Fe(OH)2 were immobilized on rGO to form FeOxH–rGO nanocomposites. The as-prepared FeOxH–rGO nanocomposites exhibited peroxidase-like catalytic activity in the H2O2-mediated oxidation of Amplex Red (AR) to fluorescent resorufin. Our AR/FeOxH–rGO probe allowed the detection of H2O2 down to 50 nM within 10 min under microwave irradiation (170 W). The catalytic activity of FeOxH–rGO was significantly suppressed in the presence of S2− because of the formation of FeS on the FeOxH–rGO nanocomposites' surfaces. The H2O2/AR–FeOxH–rGO probe provided a limit of detection (signal-to-noise ratio = 3) of 50 nM for S2− with high selectivity (>100-fold) with respect to other anions. Taking advantage of their high stability and selectivity, we employed our H2O2/AR–FeOxH–rGO probe for the detection of S2− in hot spring samples (75.1–619.5 μM) and the results showed good correlation (r = 0.98) with results from inductively coupled plasma mass spectrometry. This label-free, rapid, and simple sensing system shows great potential for the detection of S2− ions in real samples.
1 Introduction
Natural enzymes possess high substrate specificity and high catalytic efficiency, and have been extensively investigated in many applications such as medicine, environmental analysis, and food processing.1 However, the catalytic activities of natural enzymes are easily affected by environmental conditions such as pH, temperature, ionic strength, surfactants, and organic solvents.2 In recent years, great attention has been paid to the construction and discovery of novel enzyme mimetic nanomaterials, specifically bimetallic nanoparticles (NPs) and hybrid nanomaterials.3–5 For example, it has been found that many noble metal-based NPs, including AuBi, AuPt, AuHg, AuPb, AgAu, and AgPt bimetallic alloy NPs, exhibit high catalytic activity.3 The enzyme-like activity (oxidase, peroxidase, and catalase) of Au NPs can be tuned by reaction of different metal ions.4 For example, Au NPs in the presence of Bi3+, Ag+, and Hg2+ exhibit peroxidase-, oxidase-, and catalase-like activities by forming AuBi, AuAg, and AuHg alloy nanolayers on particle surfaces, respectively.4 Furthermore, it has been demonstrated that graphene-based carbon materials promote electron transfer between the substrate and catalytic NPs, and improve their dispersibility.5 Many graphene-supported metal NPs or metal oxide NP hybrid nanomaterials, including Au@Pd nanoparticle–graphene hybrids, graphene oxide–Fe3O4 magnetic nanocomposites, Co3O4-reduced graphene oxide (rGO) nanocomposites, and CoFe2O4 immobilized on rGO nanocomposites, have been shown to act as peroxidase mimics for H2O2-mediated reactions.6 Furthermore, these graphene-based hybrid nanomaterials have been employed for the detection of glucose and DNA, and cancer cells and degradation of dyes.6,7
The sulfide anion (S2−) is a traditional toxic pollutant found in water owing to not only industrial wastewater but also microbial reduction of sulfate by anaerobic bacteria and the sulfur-containing amino acids in meat proteins.8 Once protonated, H2S is even more toxic than the sulfide itself. Continuous and high concentration exposure to H2S can cause various physiological and biochemical problems such as Down's syndrome, Alzheimer's disease, diabetes, respiratory paralysis, and liver cirrhosis.9 Thus, a rapid and sensitive method for the detection of S2− is essential for environmental protection, clinical diagnostics, and microbial infestations. So far, many methods have been employed for the determination of S2− concentrations, including titration, spectroscopy, electrochemistry, chromatography, and combinations thereof.10 However, these probing systems are time-consuming, complicated procedures, requiring large sample volumes and specialized skills. Thus, there is a need to develop sensitive and simple probes not only for qualitative analysis but also for determination of S2− in real samples at trace levels.
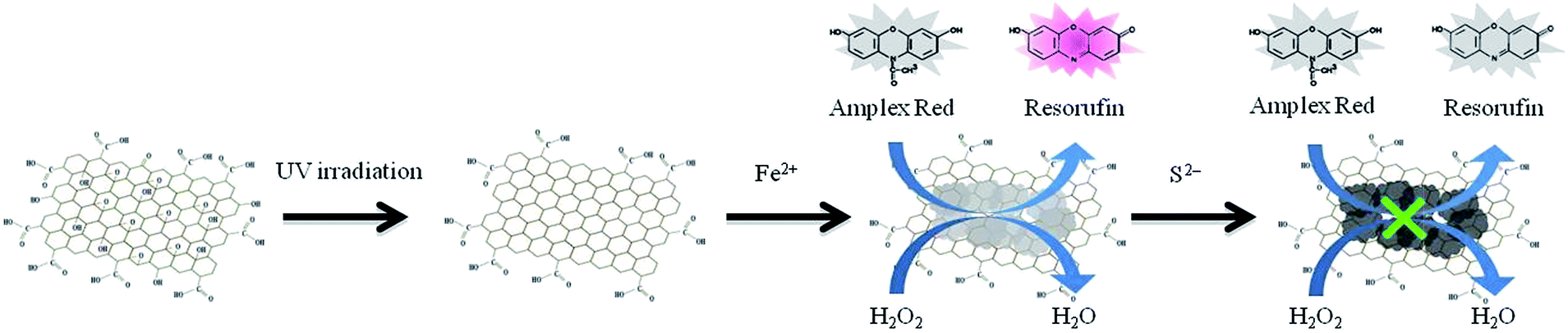
In this study, we immobilized iron hydroxide/oxide on rGO from rGO (size ∼ 300 nm; prepared from irradiation of GO with UV light for 5 h) and iron ions (Fe2+) in 5.0 mM Tris–borate solution (pH 7.0). The iron(III) oxide-hydroxide [FeO(OH)] and iron(II) hydroxide [Fe(OH)2] were immobilized on rGO to form FeOxH–rGO nanocomposites. The FeOxH–rGO nanocomposites exhibited high catalytic activity for the H2O2-mediated oxidation of Amplex Red (AR; 10-acetyl-3,7-dihydroxyphenoxazine) to fluorescent resorufin (7-hydroxy-3H-phenoxazin-3-one) (Scheme 1).11 To demonstrate the practicality of FeOxH–rGO, the as-prepared FeOxH–rGO nanocomposites were first employed for the rapid detection of H2O2 assisted with microwave irradiation. We further applied the AR/H2O2–FeOxH–rGO system for the sensing of S2− based on the analyte-induced inhibition of the catalytic activity of FeOxH–rGO nanocomposites (Scheme 1). The practicality of this approach was validated through the detection of S2− in stream water, lake water, tap water, and hot springs.
 |
| | Scheme 1 Schematic representation of the preparation of peroxidase-like FeOxH–rGO nanocomposites for the detection of sulfide ions based on the inhibition of enzymatic activity. | |
2 Experimental
2.1 Chemicals
Tris(hydroxymethyl)aminomethane (Tris), hydrochloric acid, boric acid, and all metal salts used in this study were purchased from Mallinckrodt Baker (Phillipsburg, NJ, USA). AR was purchased from Invitrogen (Eugene, Oregon, USA). Sodium cyanide, sodium thiocyanate, sodium acetate, sodium bromide, sodium chloride, sodium carbonate, sodium iodide, sodium nitrate, sodium phosphate, potassium permanganate, sodium sulfide, and graphite (7–11 μm) were obtained from Alfa Aesar (Ward Hill, MA, USA). Hydrogen peroxide was purchased from SHOWA (Tokyo, Japan). Sulfuric acid and phosphoric acid were purchased from J. T. Baker (Phillipsburg, NJ, USA). Milli-Q ultrapure water (Millipore, Billerica, MA, USA) was used in all experiments. The buffer used in this study was a solution of Tris–borate (50 mM, pH 7.0 adjusted Tris with 200 mM boric acid).
2.2 Preparation of FeOxH–rGO
GO was synthesized using an improved Hummers' method.12 Briefly, a mixture of graphite flakes (0.75 g) and KMnO4 (4.5 g) was added to a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of concentrated H2SO4 and H3PO4 (100 mL). The mixture was then heated to 50 °C and stirred for 12 h. The reaction was cooled to room temperature in an ice bath, and then poured into 100 mL of deionized (DI) water containing 3 mL of 30% H2O2. The aqueous mixture was then centrifuged at a relative centrifugal force of 35000g for 1 h, and the supernatant was decanted. The remaining pellet was repeatedly washed with 200 mL of DI water until the washings reached a pH of 7.0. The aqueous solution was then sonicated for 1 h and centrifuged at a relative centrifugal force of 25000g for 0.5 h. The GO solution was collected and the remaining pellet was discarded. The GO concentration in the supernatant was determined to be ∼1.2 g L−1 (denoted as the 100× concentration for simplicity) using the freeze-drying method. The reduced graphene oxide (rGO) was prepared from irradiation of 10× GO in 5.0 mM Tris–borate solution (pH 7.0) with a hand-held UV lamp (365 nm; 140 mW cm−2) for 5 h. For preparation of FeOxH–rGO, FeCl2 (100 μM) was mixed with GO (1×) in a Tris–borate solution (5.0 mM, pH 7.0) and reacted for 1 h. The resulting FeO(OH) and Fe(OH)2 were immobilized on rGO to form the FeOxH–rGO nanocomposites.
:1 mixture of concentrated H2SO4 and H3PO4 (100 mL). The mixture was then heated to 50 °C and stirred for 12 h. The reaction was cooled to room temperature in an ice bath, and then poured into 100 mL of deionized (DI) water containing 3 mL of 30% H2O2. The aqueous mixture was then centrifuged at a relative centrifugal force of 35000g for 1 h, and the supernatant was decanted. The remaining pellet was repeatedly washed with 200 mL of DI water until the washings reached a pH of 7.0. The aqueous solution was then sonicated for 1 h and centrifuged at a relative centrifugal force of 25000g for 0.5 h. The GO solution was collected and the remaining pellet was discarded. The GO concentration in the supernatant was determined to be ∼1.2 g L−1 (denoted as the 100× concentration for simplicity) using the freeze-drying method. The reduced graphene oxide (rGO) was prepared from irradiation of 10× GO in 5.0 mM Tris–borate solution (pH 7.0) with a hand-held UV lamp (365 nm; 140 mW cm−2) for 5 h. For preparation of FeOxH–rGO, FeCl2 (100 μM) was mixed with GO (1×) in a Tris–borate solution (5.0 mM, pH 7.0) and reacted for 1 h. The resulting FeO(OH) and Fe(OH)2 were immobilized on rGO to form the FeOxH–rGO nanocomposites.
2.3 Characterization
Transmission electron microscope (TEM) images of the rGO and FeOxH–rGO nanocomposites were recorded using a Hitachi H7100 TEM, operated at 75 kV. Samples for TEM and energy-dispersive X-ray spectroscopy (EDS) measurements were prepared by placing aliquots (20 μL) of the rGO or FeOxH–rGO solutions on a carbon-coated copper grid (copper 200 mesh). After standing for 2 h at ambient temperature, the solution of rGO or FeOxH–rGO was removed. EDS analysis of FeOxH–rGO using a 0.7 nm diameter electron probe was employed to determine their chemical identities. X-ray diffraction (XRD) samples were prepared by depositing FeOxH–rGO on a Si(100) wafer, and XRD measurements were performed at room temperature using a Rigaku 18 kW rotating anode source X-ray diffractometer (The Woodlands, Texas, USA) with the Cu Kα1 line (λ = 1.54 Å, energy = 8.8 keV) operated at 50 kV, 100 mA, and slits set at 10 × 2 mm2. X-ray photoelectron spectroscopy (XPS) was performed using a VG ESCA scientific theta probe spectrometer (Uppsala, Sweden) in the constant analyzer energy mode with a pass energy of 28 eV and Al Kα (1486.6 eV) radiation as the excitation source. Raman spectra were recorded using a Raman spectrometer (DongWoo 500i; KyungGiDo, Korea) equipped with a 50× objective Nd:YAG laser (532 nm) and a charge-coupled detector. The signal collection time for each sample was 30 s. A Zetasizer 3000HS analyzer (Malvern Instruments, Malvern, UK) was used for analysis of dynamic light scattering and zeta potential of rGO and FeOxH–rGO nanocomposites.
2.4 Peroxidase-like activity assay
Aliquots (400 μL) of Tris–borate solutions (5.0 mM, pH 7.0) containing Fe2+ (125 μM), GO (1.25×), FeOxH–GO (1.25×), rGO (1.25×), or FeOxH–rGO (1.25×) were equilibrated at room temperature for 1 h. Tris–borate solution (100 μL, 5.0 mM, pH 7.0) containing AR (50 μM) and H2O2 (500 μM) was then added to each of the mixtures and left for 2 h before fluorescence measurements with excitation at 530 nm (Synergy 4 monochromatic microplate spectrophotometer, Biotek Instruments, Winooski, VT, USA).
2.5 Catalytic sensing of S2−
Aliquots (350 μL) of the Tris–borate solution (5.0 mM, pH 7.0) containing FeOxH–rGO (0.143×) were equilibrated at room temperature for 10 min. Tris–borate solution (50 μL, 5.0 mM, pH 7.0) containing S2− (0–15 μM) was separately added to each of the FeOxH–rGO nanocomposite solutions and left for an additional 30 min. Tris–borate solution (100 μL, 5.0 mM, pH 7.0) containing AR (50 μM) and H2O2 (50 μM) was then added to each of the mixtures and left for 2 h before fluorescence measurements with excitation at 530 nm.
2.6 Enzyme kinetic analysis
Kinetic measurements were conducted with a black 96-well microplate using a Synergy 4 monochromatic microplate spectrophotometer. The AR/H2O2 substrates in Tris–borate solution (180 μL, pH 7.0) were separately added to each well of a microtiter plate, and aliquots (20 μL) of peroxidase-like FeOxH–rGO nanocomposite solutions (1×) were then added to the plate. The reaction progress was monitored every 30 s for 2 h by recording the fluorescence of the reaction product, resorufin, at 585 nm with an excitation wavelength of 530 nm. Variable concentrations (0.5–25 μM) of AR with a constant H2O2 concentration (10 μM) were investigated in the catalytic reactions. In addition, variable H2O2 concentrations (100–7500 μM) with a constant AR concentration (10 μM) were also investigated.
2.7 Analysis of real samples
Water samples collected from a stream near the National Taiwan Ocean University campus, a lake on the National Taiwan University campus, and local tap water were filtered through a 0.22 μm membrane. For the detection of S2−, aliquots (300 μL) of the Tris–borate solution (5.0 mM, pH 7.0) containing FeOxH–rGO (0.167×) were equilibrated at room temperature for 10 min. The 2-fold diluted water samples were spiked with S2− (0–5.0 μM) in a Tris–borate solution (100 μL, 5.0 mM, pH 7.0) and then separately added to the FeOxH–rGO nanocomposite solutions. After reacting for 30 min, the Tris–borate solution (100 μL, 5.0 mM, pH 7.0) containing AR (50 μM) and H2O2 (50 μM) was added to each of the mixtures and left for 2 h before fluorescence measurements with excitation at 530 nm.
Hot spring water samples collected from Yangmingshan National Park were filtered through a 0.22 μm membrane. For the detection of S2−, aliquots (300 μL) of the Tris–borate solution (5.0 mM, pH 7.0) containing FeOxH–rGO (0.167×) were equilibrated at room temperature for 10 min. The 40-fold diluted hot spring water samples were prepared in a Tris–borate solution (100 μL, 5.0 mM, pH 7.0) and then separately added to the FeOxH–rGO nanocomposite solutions. After reacting for 30 min, the Tris–borate solution (100 μL, 5.0 mM, pH 7.0) containing AR (50 μM) and H2O2 (50 μM) was added to each of the mixtures and left for 2 h before fluorescence measurements with excitation at 530 nm. Moreover, all samples were analyzed by inductively coupled plasma-mass spectrometry (ICP-MS; 7700 Series, Agilent Technologies, California, USA); the samples were prepared in 2% HNO3.
3 Results and discussion
3.1 Peroxidase-like activity of FeOxH–rGO
The peroxidase-like activity of the FeOxH–rGO nanocomposites was evaluated using the typical peroxidase substrate AR in the presence of H2O2. H2O2 acted as an electron acceptor of the FeOxH–rGO nanocomposites for the catalytic oxidation of AR with a 1:1 stoichiometry, which yielded a highly fluorescent and colored product, resorufin (quantum yield: 0.83; absorption coefficient: 5.4 × 104 cm−1 M−1 at 570 nm).13 The AR was selected as the substrate based on the stability of its oxidized product (resorufin) at pH values greater than 5.0 and its high quantum yield (>80%) as well as long excitation and emission wavelengths. Moreover, fluorescence-based sensors are typically much sensitive (>100-fold) than colorimetric ones, we expected our H2O2/AR–FeOxH–rGO probe to provide a comparatively lower LOD values for S2−. In our previous study,14 we found that 3,3′,5,5′-tetramethylbenzidine (TMB) was not a suitable substrate for nanoparticles-mediated catalytic oxidation of AR in the presence of H2O2 in neutral solution, mainly because TMB could only react with H2O2 by natural peroxidase and peroxidase mimic nanoparticles at low pH values (3.0–5.0). The FeOxH–rGO nanocomposites likely catalyzed a one-electron oxidation of a nonfluorescent AR to form a nonfluorescent AR radical.15 Subsequently, these two AR radicals underwent an enzyme-independent dismutation reaction to form fluorescent resorufin. The Fe2+, GO, and rGO exhibited relatively low catalytic activity for the H2O2-mediated AR reaction (curves a, b, and d in Fig. 1A). In contrast, the FeOxH–GO and FeOxH–rGO (curves c and e in Fig. 1A) relative to GO and rGO exhibited 70- and 770-fold fluorescence intensity at 585 nm when excited at 530 nm. Our results indicated that FeOxH–rGO nanocomposites have high peroxidase-like activity. The catalytic mechanism of FeOxH–rGO nanocomposites may follow Fenton-like reactions due to Fe2+/Fe3+ in deposited FeOxH.16 When FeCl2 was prepared in Tris–borate solution (5.0 mM, pH 7.0), the iron(III) oxide-hydroxide (4Fe2+ + O2 + 6H2O → 4FeO(OH) + 8H+) and iron(II) hydroxide (Fe2+ + 2H2O → Fe(OH)2 + 2H+) were formed and immobilized on GO or rGO.16 As shown in Fig. 2B and C, the FeOxH nanostructures were randomly distributed on the surface of GO and rGO.
 |
| | Fig. 1 (A) Fluorescence spectra of 5.0 mM Tris–borate (pH 7.0) containing AR (10 μM) and H2O2 (100 μM) in the presence of (a) Fe2+ (100 μM), (b) GO (1×), (c) FeOxH–GO [prepared from GO (1×) and Fe2+ (100 μM) in Tris–borate solution], (d) rGO (1×), and (e) FeOxH–rGO [prepared from rGO (1×) and Fe2+ (100 μM) in Tris–borate solution]. (B) UV-vis absorption spectra of 5.0 mM Tris–borate (pH 7.0) in the presence of (a) GO, (b) FeOxH–GO, (c) rGO, and (d) FeOxH–rGO. Inset to (A): photograph of the fluorescence of the solutions upon excitation under a hand-held UV lamp (365 nm). The fluorescence intensity (IF) and absorption (Abs) are plotted in arbitrary units (a. u.). The excitation wavelength in (A) was set at 530 nm. | |
 |
| | Fig. 2 (A–C) Low-magnification TEM images of 5.0 mM Tris–borate (pH 7.0) containing (A) rGO (1×), (B) FeOxH–GO (1×), and (C) FeOxH–rGO (1×) and (D and E) high-magnification TEM images of 5.0 mM Tris–borate (pH 7.0) containing (D) FeOxH–GO (1×) and (E) FeOxH–rGO (1×). Other conditions were the same as those described in Fig. 1. | |
The Raman spectra of FeOxH–rGO revealed that the FeO(OH) and Fe(OH)2 species were dominant on rGO (Fig. S1, ESI†). The peaks at 210, 272, 384, and 995 cm−1 were assigned to FeO(OH), while the bands at 580 cm−1 were attributed to Fe(OH)2. In addition, the atomic ratio of O to Fe of bare FeOxH was evaluated by the EDS measurement to be about 1.98:1, consistent with that of FeOOH or Fe(OH)2 (Fig. S2, ESI†). The XPS measurement further revealed the FeO(OH) (43.2%) and Fe(OH)2 (50.4%) are the major species in the FeOxH–rGO nanocomposite (Fig. S3, ESI†). Moreover, no crystalline FeOxH was found in the XRD image (data not shown), suggesting that the deposited FeO(OH) and Fe(OH)2 are amorphous. Recently, amorphous FeOOH and Fe(OH)2 nanostructures have been employed as catalysts for photoelectrochemical water splitting, electrode materials for lithium-ion batteries, and degradation of dye pollutants.17,18 To our knowledge, however, amorphous FeOOH and Fe(OH)2 used as enzyme-like materials in solution have not been attempted, although its specific surface area is higher than crystalline iron oxide (such as Fe2O3 and Fe3O4). FeOOH and Fe(OH)2 are merely used as catalysts in homogenous aqueous solution, probably because their nanostructures are difficult to confine; they easily exist as gel-like structures suspended in the solution. Another disadvantage of amorphous FeOOH and Fe(OH)2 is their tendency to form crystalline iron oxides or dissolve in preparation or storage, which may greatly reduce their catalytic activity as their surface area is greatly diminished. We noted that our rGO-supported FeO(OH) and Fe(OH)2 were stable in aqueous solution at room temperature for at least two months. This phenomenon is presumably due to the strong association of FeO(OH) and Fe(OH)2 with rGO, improving their stability.
We noted that the peroxidase-like activity of FeOxH–rGO (curve e in Fig. 1) was about 10-fold higher than that of FeOxH–GO (curve c in Fig. 1). This may be attributed to the fact that rGO has a stronger adsorption ability to AR and higher conductivity.19 The UV-vis absorption spectra of GO show a broad absorption band with a shoulder in the UV region. The absorption band (230 nm) was attributed to the π → π* transition of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in the sp2 hybrid region. The shoulder at ∼300 nm was caused by the n → π* electronic transition of peroxide and/or epoxide functional groups in GO.20 The slightly stronger absorption of rGO indicated that some oxygen-containing carbons were reduced to CC, which can provide more π orbitals for adsorption of AR molecules via π–π stacking and transfer of electrons to oxidize AR. Relative to GO, the higher ratio of CC/C–C (86.2% versus 56.9%; Fig. S4, ESI†) of rGO further supports our reasoning. We also noted that at constant concentrations of AR (500 nM) and GO or rGO (1×), about 70% and 95% of AR molecules were adsorbed on GO and rGO, respectively. It has been reported that enzyme-mimicking nanoparticles transfer electrons between pairs of different oxidation states of metal ions to drive their catalytic activity.3–5 Therefore, the various valence states of Fe2+/Fe3+ on particle surfaces and high conductivity of rGO accounted for the nanocomposites' high peroxidase-like activity.
C bond in the sp2 hybrid region. The shoulder at ∼300 nm was caused by the n → π* electronic transition of peroxide and/or epoxide functional groups in GO.20 The slightly stronger absorption of rGO indicated that some oxygen-containing carbons were reduced to CC, which can provide more π orbitals for adsorption of AR molecules via π–π stacking and transfer of electrons to oxidize AR. Relative to GO, the higher ratio of CC/C–C (86.2% versus 56.9%; Fig. S4, ESI†) of rGO further supports our reasoning. We also noted that at constant concentrations of AR (500 nM) and GO or rGO (1×), about 70% and 95% of AR molecules were adsorbed on GO and rGO, respectively. It has been reported that enzyme-mimicking nanoparticles transfer electrons between pairs of different oxidation states of metal ions to drive their catalytic activity.3–5 Therefore, the various valence states of Fe2+/Fe3+ on particle surfaces and high conductivity of rGO accounted for the nanocomposites' high peroxidase-like activity.
3.2 Effect of irradiation
We demonstrated that rGO plays an important role in enhancing the catalytic activity of FeOxH. Fig. 3 shows that the catalytic activity of FeOxH–rGO nanocomposites was increased with increasing UV-irradiation time in the preparation of rGO. The fluorescence intensity of resorufin at 585 nm increased initially on increasing the irradiation time before reaching a plateau at ∼5 h. This result is consistent with the UV-vis absorption of rGO, which was prepared from GO with different UV-irradiation times (0–10 h; Fig. S5, ESI†). The color of GO (rGO) solutions changed from light brown to dark black and absorbance became stronger with increasing UV-irradiation time, from 0 to 10 h. Under optimized conditions for the preparation of rGO (irradiation of GO with UV light for 5 h), our AR–FeOxH–rGO probe allowed detection of H2O2 concentrations down to 1.0 μM under the catalytic reaction time of 2 h (curve a in Fig. S6, ESI†). To shorten the analysis time (<10 min), we employed microwave irradiation to aid the catalytic reaction. Microwave heating is one type of electroheating technique that utilizes specific wavelengths of electromagnetic energy.21 When applying microwave irradiation to metallic and metal oxide nanomaterials, the electric and magnetic components change rapidly, and the molecules cannot respond quickly to the change in direction, giving rise to friction and therefore causing them to quickly warm up.22 The acceleration of the catalytic reaction rate of H2O2/AR–FeOxH–rGO under microwave irradiation is probably due to the superheating effect produced in a microwave field.23 The heat and electron transfer of FeOxH–rGO was strongly influenced after GO interacted with the microwave field.24 It has been demonstrated that metal oxide–rGO nanocomposites have better dielectric constants due to the significantly increased conductivity from rGO.24 Metal oxides incorporated with rGO could have enhanced microwave-absorbing properties.25 In addition, the different dielectric properties of the liquid and FeOxH–rGO nanocomposites might result in localized temperature differences, creating strong convection currents at the surface of the microwaved FeOxH–rGO nanocomposites.24,25 Therefore, diffusions of the reaction products were rapidly promoted away from the surface. Under the assistance of microwave irradiation (170 W), our AR–FeOxH–rGO probe allowed the detection of H2O2 with a limit of detection (LOD; signal-to-noise (S/N) ratio = 3) of ∼50 nM within 10 min (curve b in Fig. S5, ESI†). One of the possible factors for this ultrahigh sensitivity for H2O2 is the microwave temperature; under microwave irradiation of 170 W, the reaction temperature was raised to ∼75 °C. However, we noted that the reaction time needed to reach completion was 2 h even at 75 °C (data not shown). In another control experiment, we noted the microwave irradiation caused negligible fluorescence change to 5.0 mM Tris–borate (pH 7.0) solution containing AR (10 μM)–H2O2 (10 μM). This microwave-assisted catalytic reaction not only shortened the analysis time to 10 min, but also provided near one order of magnitude greater sensitivity than the above results. Compared with other GO-based nanocomposites with peroxidase-like activities, the preparation of FeOxH–rGO is relatively simple and cost-effective and the AR–FeOxH–rGO probe shows comparable sensitivity for H2O2 detection.6,7
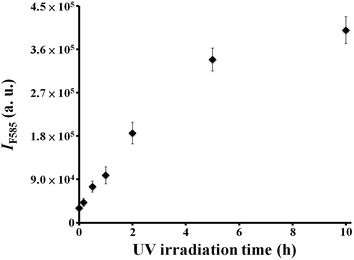 |
| | Fig. 3 Fluorescence response (IF585) of 5.0 mM Tris–borate (pH 7.0) containing AR (10 μM), H2O2 (100 μM), and FeOxH–rGO [prepared from Fe2+ (100 μM) and UV-irradiated (0–10 h) GO (1×)]. Error bars represent the standard deviations from three repeated experiments. The fluorescence intensities at 585 (IF585) are plotted in arbitrary units (a. u.). Other conditions were the same as those described in Fig. 1. | |
3.3 Sensing of sulfide
Fig. 4A (curve b) reveals the poorly developed fluorescence intensity of the H2O2/AR–FeOxH–rGO (0.1×) system in the presence of S2− (10 μM) in Tris–borate solution (5.0 mM, pH 7.0). The low catalytic activity of FeOxH–rGO in the presence of S2− is presumably because of the formation of FeS and Fe2S3 on the FeOxH–rGO due to the strong affinity of S2− for surface iron ions on nanocomposites Fe2+ (Ksp (FeS) ∼ 6 × 10−19) and Fe3+ (Ksp (Fe2S3) ∼ 1 × 10−88). The formation of FeS and Fe2S3 may block the active sites of FeOxH–rGO and diminish their peroxidase-like activity. The EDS (Fig. 4B) and Raman spectra (Fig. 4C) further supported that sulfide was deposited on FeOxH–rGO. In addition, we used ICP-MS to quantify that about 95% of S2− (10 μM) ions were binding to FeOxH–rGO (0.1×). According to the Michaelis–Menten equation (1/ν = KM/vmax (1/[S] + 1/KM)), the kinetic data, including the Michaelis constant (KM) and maximal velocity (vmax) of FeOxH–rGO nanocomposites in the absence and presence of S2−, were calculated and are listed in Table 1. From the double reciprocal plots of catalytic velocity against one of the substrate concentrations when the other substrate was fixed at three concentration levels, we demonstrated that the catalytic reaction of H2O2/AR–FeOxH–rGO followed a ping-pong mechanism because the slopes of the lines were parallel (Fig. S7, ESI†).26 This result revealed that, like HRP, the FeOxH–rGO nanocomposites bind and react with the first substrate (AR or H2O2) and then release the first product before reacting with the other substrate. KM is often associated with the affinity of the catalyst NPs for the substrates. By comparing the apparent kinetic parameters, the KM value of FeOxH–rGO nanocomposites in the presence of S2− with AR as the substrate was slightly higher than that in the absence of S2−, revealing that FeOxH–rGO nanocomposites have a lower affinity with AR when FeS and/or Fe2S3 were deposited on their surfaces. The KM value of FeOxH–rGO nanocomposites in the presence of S2− with H2O2 as the substrate was slightly lower than that for FeOxH–rGO nanocomposites alone, which agrees with reports that FeS and Fe2S3 have a strong affinity with H2O2.27 Although FeOxH–rGO nanocomposites in the presence of S2− have a lower KM for H2O2, the ∼2.5-fold lower vmax value indicated that the deposited FeS and Fe2S3 did not promote the catalytic reaction.
 |
| | Fig. 4 (A) Fluorescence spectra of 5.0 mM Tris–borate (pH 7.0) containing AR (10 μM), H2O2 (10 μM), and FeOxH–rGO (0.1X) in the (a) absence and (b) presence of S2− (10 μM). (B) EDS and (C) Raman spectra of FeOxH–rGO (10X) in the presence of S2− (1.0 mM). | |
Table 1 Comparison of the apparent Michaelis constant (KM) and maximal velocity (vmax) between FeOxH–rGO nanocomposites (0.1×) in the absence and presence of S2− (10 μM)
| Catalyst |
Substrate |
KM (μM) |
vmax (μM s−1) |
| FeOxH–rGO |
AR |
3.67 |
1.3 × 10−3 |
| FeOxH–rGO + S2− |
AR |
5.16 |
7.1 × 10−4 |
| FeOxH–rGO |
H2O2 |
326 |
5.6 × 10−4 |
| FeOxH–rGO + S2− |
H2O2 |
166 |
2.2 × 10−4 |
We further investigated the selectivity and sensitivity of the H2O2 (10 μM)/AR (10 μM)–FeOxH–rGO (0.1×) probe for sensing S2−. The catalytic activity of FeOxH–rGO was significantly reduced by S2− at room temperature, when compared with other tested anions, including CH3COO−, PO43−, S2O32−, SO42−, NO3−, Cl−, Br−, I−, NO2−, CN−, SCN−, AsO2−, and AsO43− (Fig. 5A, the concentration of each anion was 10 μM). In addition, tolerance concentrations of the other anions (within a relative error of ±5%) during the sensing of S2− (10 μM) with the H2O2/AR–FeOxH–rGO probe were at least 100 times higher than that of the S2− (Fig. S8, ESI†). The fluorescence response of the H2O2/AR–FeOxH–rGO probe decreased on increasing the concentration of S2− ions (Fig. 5B). We obtained a linear response in the plot of the expression (IF0 − IF)/IF0 against the concentration of S2− over the range 0.1–1.5 μM (r = 0.99), where IF0 and IF represent the fluorescence intensities of the mixtures in the absence and presence, respectively, of the added S2−. The H2O2/AR–FeOxH–rGO probe provided an LOD for S2− ions (S/N = 3) of ∼50 nM. This LOD for S2− was comparable to those using other optical sensors with functional nanoparticles.28,29 Although the sensitivities of some chemosensors are higher than the H2O2/AR–FeOxH–rGO probe, those chemosensors require complicated and multistep complicated synthesis and use of sophisticated equipment.30
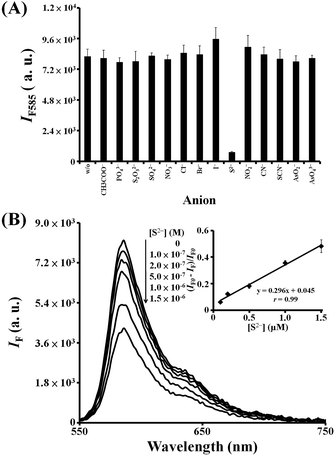 |
| | Fig. 5 (A) Selectivity of the H2O2/AR–FeOxH–rGO probe toward S2− ions. Fluorescence response (IF585) of 5.0 mM Tris–borate solution (pH 7.0) containing AR (10 μM), H2O2 (10 μM), and FeOxH–rGO nanocomposites (0.1×) in the absence or presence of anions (10 μM) at 585 nm. (B) Validation of the use of H2O2/AR–FeOxH–rGO probe for the detection of S2− (0–1.5 μM). The inset to (B): values of (IF0 − IF)/IF0 were plotted against S2− concentration. IF0 and IF represent the fluorescence intensities of the solutions at 585 nm in the absence and presence of S2−, respectively. Error bars in the inset represent the standard deviations from three repeated experiments. Other conditions were the same as those described in Fig. 4. | |
3.4 Detection of S2− in real samples
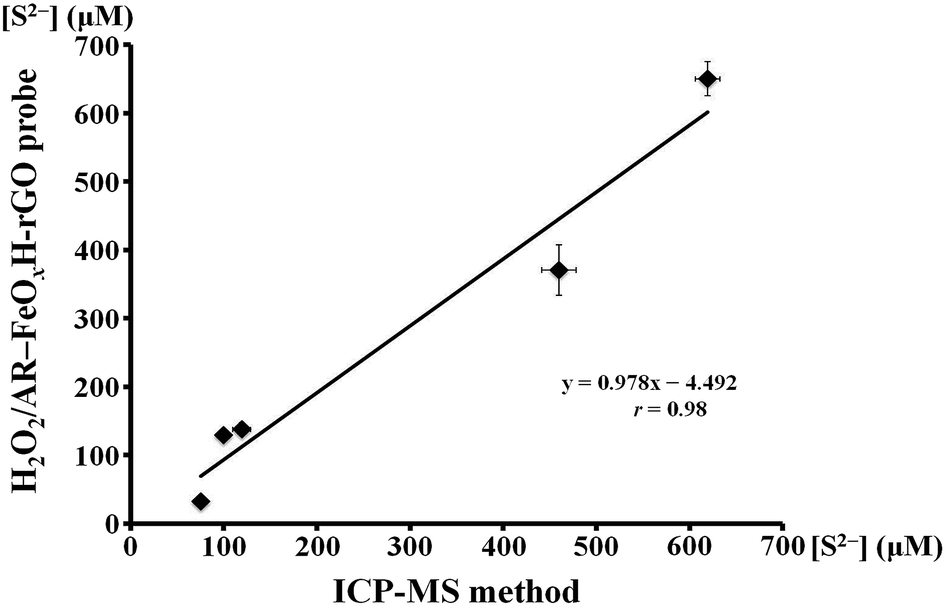
To validate that our proposed sensing strategy could have practical application for S2− analysis in water samples, we applied the H2O2/AR–FeOxH–rGO sensor to determine the levels of S2− in stream, lake, and tap water samples. Before analysis, each of the three samples were filtered through a 0.22 μm membrane and diluted 10-fold in 5.0 mM Tris–borate solution (pH 7.0). Here, we obtained linear correlations (r = 0.98–0.99) between the relative fluorescence changes ((IF0 − IF)/IF0) and the concentration of spiked S2− (Fig. S9, ESI†), where IF0 and IF represent the fluorescence intensities of the mixtures in the absence and presence, respectively, of the spiked S2−. In these measurements, the probe provided recoveries of 104.2–107.3% for S2− ions (0.5 μM). The minimum concentration of S2− ions detectable by our H2O2/AR–FeOxH–rGO probe in these water samples was ∼100 nM. Neither an ICP-MS-based system nor our sensing approach could detect the presence of any S2− ions in these original water samples. We further applied the H2O2/AR–FeOxH–rGO probe to determine the S2− ions in five sulfur spring waters collected from Yangmingshan National Park (Taipei, Taiwan). Fig. 6 shows the good linear correlation (r = 0.98) between the results obtained using the H2O2/AR–FeOxH–rGO probe and ICP-MS over concentrations ranging from 75.1 to 619.5 μM, suggesting that our probe is useful for screening S2− concentrations in hot spring waters. Therefore, the proposed methods are applicable for practical analysis of S2− in environmental samples.
 |
| | Fig. 6 Comparison between ICP-MS and the H2O2/AR–FeOxH–rGO probe for detection of S2− in hot spring water samples. There was a linear correlation between the S2− concentrations in five hot spring waters determined using ICP-MS and H2O2/AR–FeOxH–rGO assays. Error bars represent the standard deviations from three repeated experiments. | |
4 Conclusions
In summary, nanocomposites of amorphous FeOxH immobilized on rGO (FeOxH–rGO) were successfully synthesized by the simple reaction of GO partially reduced by ultraviolet irradiation with Fe2+ in aqueous solution. We demonstrated that FeOxH–rGO nanocomposites exhibit intrinsic peroxidase-like activity. With the assistance of microwave irradiation, our AR/FeOxH–rGO probe allowed detection of H2O2 down to 50 nM within 10 min. In the presence of S2−, the catalytic activity of FeOxH–rGO became lower because the formation of FeS and Fe2S3 may block the active sites of FeOxH–rGO. The H2O2/AR–FeOxH–rGO probe provided an LOD of 50 nM for S2− with high selectivity (>100-fold). Owing to the high stability and selectivity of the nanocomposites, the H2O2/AR–FeOxH–rGO probe allowed the detection of S2− in hot spring waters (75.1–619.5 μM) and the results showed good correlation (r = 0.98) with ICP-MS. This label-free, low cost, and rapid nanosensor holds great potential for screening S2− in real water samples.
Acknowledgements
This study was supported by the Ministry of Science and Technology of Taiwan under contracts NSC 101-2628-M-019-001-MY3, 102-2113-M-019-001-MY3 and 102-2627-M-019-001-MY3.
Notes and references
-
(a) L. M. Podust and D. H. Sherman, Nat. Prod. Rep., 2012, 29, 1251–1266 RSC;
(b) J. A. Gerlt and P. C. Babbitt, Curr. Opin. Chem. Biol., 2009, 13, 10–18 CrossRef CAS PubMed;
(c) R. Masella, R. Di Benedetto, R. Varì, C. Filesi and C. Giovannini, J. Nutr. Biochem., 2005, 16, 577–586 CrossRef CAS PubMed.
-
(a) M. Naushad, Z. A. ALOthman, A. B. Khan and M. Ali, Int. J. Biol. Macromol., 2012, 51, 555–560 CrossRef CAS PubMed;
(b) P. V. Iyer and L. Ananthanarayan, Process Biochem., 2008, 43, 1019–1032 CrossRef CAS PubMed;
(c) C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS PubMed;
(d) G. Feller, J. Phys.: Condens. Matter, 2010, 22, 323101 CrossRef PubMed;
(e) S. Yabuki, Anal. Sci., 2014, 30, 213–217 CrossRef CAS.
-
(a) C.-W. Lien, C.-C. Huang and H.-T. Chang, Chem. Commun., 2012, 48, 7952–7954 RSC;
(b) W. He, Y. Liu, J. Yuan, J.-J. Yin, X. Wu, X. Hu, K. Zhang, J. Liu, C. Chen, Y. Ji and Y. Guo, Biomaterials, 2011, 32, 1139–1147 CrossRef CAS PubMed;
(c) C.-I. Wang, C.-C. Huang, Y.-W. Lin, W.-T. Chen and H.-T. Chang, Anal. Chim. Acta, 2012, 745, 124–130 CrossRef CAS PubMed;
(d) C.-I. Wang, W.-T. Chen and H.-T. Chang, Anal. Chem., 2012, 84, 9706–9712 CrossRef CAS PubMed;
(e) H. You, Z. Peng, J. Wu and H. Yang, Chem. Commun., 2011, 47, 12595–12597 RSC;
(f) Y. Chen, H. Cao, W. Shi, H. Liu and Y. Huang, Chem. Commun., 2013, 49, 5013–5015 RSC.
- C.-W. Lien, Y.-C. Chen, H.-T. Chang and C.-C. Huang, Nanoscale, 2013, 5, 8227–8234 RSC.
-
(a) J. Zhu, M. Chen, Q. He, L. Shao, S. Wei and Z. Guo, RSC Adv., 2013, 3, 22790–22824 RSC;
(b) B. F. MacHado and P. Serp, Catal. Sci. Technol., 2012, 2, 54–75 RSC;
(c) G. Blanita and M. D. Lazar, Micro Nanosyst., 2013, 5, 138–146 CrossRef CAS;
(d) Y. Liang, Y. Li, H. Wang and H. Dai, J. Am. Chem. Soc., 2013, 135, 2013–2036 Search PubMed.
-
(a) H. Chen, Y. Li, F. Zhang, G. Zhang and X. Fan, J. Mater. Chem., 2011, 21, 17658–17661 RSC;
(b) Y.-L. Dong, H.-G. Zhang, Z. U. Rahman, L. Su, X.-J. Chen, J. Hu and X.-G. Chen, Nanoscale, 2012, 4, 3969–3976 RSC;
(c) J. Xie, H. Cao, H. Jiang, Y. Chen, W. Shi, H. Zheng and Y. Huang, Anal. Chim. Acta, 2013, 796, 92–100 CrossRef CAS PubMed;
(d) J. Hao, Z. Zhang, W. Yang, B. Lu, X. Ke, B. Zhang and J. Tang, J. Mater. Chem. A, 2013, 1, 4352–4357 RSC.
-
(a) G. Nie, L. Zhang, X. Lu, X. Bian, W. Sun and C. Wang, Dalton Trans., 2013, 42, 14006–14013 RSC;
(b) X. Chen, X. Tian, B. Su, Z. Huang and M. Oyama, Dalton Trans., 2014, 43, 7449–7454 RSC;
(c) Y. Ye, T. Kong, X. Yu, Y. Wu, K. Zhang and X. Wang, Talanta, 2012, 89, 417–421 CrossRef CAS PubMed;
(d) M. I. Kim, M. S. Kim, M.-A. Woo, Y. Ye, K. S. Kang, J. Lee and H. G. Park, Nanoscale, 2014, 6, 1529–1536 Search PubMed;
(e) Y. Tao, Y. Lin, Z. Huang, J. Ren and X. Qu, Adv. Mater., 2013, 25, 2594–2599 CrossRef CAS PubMed;
(f) L.-N. Zhang, H.-H. Deng, F.-L. Lin, X.-W. Xu, S.-H. Weng, A.-L. Liu, X.-H. Lin, X.-H. Xia and W. Chen, Anal. Chem., 2014, 86, 2711–2718 CrossRef CAS PubMed;
(g) L. Deng, C. Chen, C. Zhu, S. Dong and H. Lu, Biosens. Bioelectron., 2014, 52, 324–329 CrossRef CAS PubMed;
(h) Z. Zhang, J. Hao, W. Yang, B. Lu, X. Ke, B. Zhang and J. Tang, ACS Appl. Mater. Interfaces, 2013, 5, 3809–3815 CrossRef CAS PubMed;
(i) Z. Xing, J. Tian, A. M. Asiri, A. H. Qusti, A. O. Al-Youbi and X. Sun, Biosens. Bioelectron., 2014, 52, 452–457 CrossRef CAS PubMed.
-
(a) C. L. Corkhill and D. J. Vaughan, Appl. Geochem., 2009, 24, 2342–2361 CrossRef CAS PubMed;
(b) M. J. Hawkesford and L. J. De Kok, Plant, Cell Environ., 2006, 29, 382–395 CrossRef CAS PubMed;
(c) E. Pouokam, J. Steidle and M. Diener, Biol. Pharm. Bull., 2011, 34, 789–793 CrossRef CAS;
(d) P. F. Lito, J. P. S. Aniceto and C. M. Silva, Water, Air, Soil Pollut., 2012, 223, 6133–6155 CrossRef CAS.
-
(a) Q. Li and J. R. Lancaster Jr, Nitric Oxide Biol. Chem., 2013, 35, 21–34 CrossRef CAS PubMed;
(b) D. J. Polhemus and D. J. Lefer, Circ. Res., 2014, 114, 730–737 CrossRef CAS PubMed;
(c) S. Mani, A. Untereiner, L. Wu and R. Wang, Antioxid. Redox Signaling, 2014, 20, 805–817 CrossRef CAS PubMed;
(d) G. K. Kolluru, X. Shen, S. C. Bir and C. G. Kevil, Nitric Oxide Biol. Chem., 2013, 35, 5–20 CrossRef CAS PubMed.
-
(a) S. K. Pandey, K.-H. Kim and K.-T. Tang, TrAC, Trends Anal. Chem., 2012, 32, 87–99 CrossRef CAS PubMed;
(b) P. Nagy, Z. Pálinkás, A. Nagy, B. Budai, I. Tóth and A. Vasas, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 876–891 CrossRef CAS PubMed;
(c) X. Hu and B. Mutus, Rev. Anal. Chem., 2013, 32, 247–256 CAS;
(d) V. S. Lin and C. J. Chang, Curr. Opin. Chem. Biol., 2012, 16, 595–601 CrossRef CAS PubMed;
(e) H. Peng, W. Chen, S. Burroughs and B. Wang, Curr. Org. Chem., 2013, 17, 641–653 CrossRef CAS.
- M. Zhou, Z. Diwu, N. Panchuk-Voloshina and R. P. Haugland, Anal. Biochem., 1997, 253, 162–168 CrossRef CAS PubMed.
-
(a) W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef;
(b) D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- B. Zhao, F. A. Summers and R. P. Mason, Free Radical Biol. Med., 2012, 53, 1080–1087 CrossRef CAS PubMed.
- C.-W. Lien, Y.-T. Tseng, C.-C. Huang and H.-T. Chang, Anal. Chem., 2014, 86, 2065–2072 CrossRef CAS PubMed.
-
(a) J. V. Rodrigues and C. M. Gomes, Free Radical Biol. Med., 2010, 49, 61–66 CrossRef CAS PubMed;
(b) H. H. Gorris and D. R. Walt, J. Am. Chem. Soc., 2009, 131, 6277–6282 CrossRef CAS PubMed.
-
(a) S. Wang, Dyes Pigm., 2008, 76, 714–720 CrossRef CAS PubMed;
(b) E. G. Garrido-Ramírez, B. K. G. Theng and M. L. Mora, Appl. Clay Sci., 2010, 47, 182–192 CrossRef PubMed;
(c) J. V. Coelho, M. S. Guedes, R. G. Prado, J. Tronto, J. D. Ardisson, M. C. Pereira and L. C. A. Oliveira, Appl. Catal., B, 2014, 144, 792–799 CrossRef CAS PubMed;
(d) N. A. Zubir, C. Yacou, J. Motuzas, X. Zhang and J. C. Diniz da Costa, Sci. Rep., 2014, 4, 4594 Search PubMed.
-
(a) F. Peng, T. Luo, L. Qiu and Y. Yuan, Mater. Res. Bull., 2013, 48, 2180–2185 CrossRef CAS PubMed;
(b) K. Zhang, V. Dwivedi, C. Chi and J. Wu, J. Hazard. Mater., 2010, 182, 162–168 CrossRef CAS PubMed;
(c) M.-L. Chen, L.-M. Shen, S. Chen, H. Wang, X.-W. Chen and J.-H. Wang, J. Mater. Chem. B, 2013, 1, 2582–2589 RSC;
(d) G. Huang, C. Zhang, Y. Long, J. Wynn, Y. Liu, W. Wang and J. Gao, Nanotechnology, 2013, 24, 395601 CrossRef PubMed;
(e) Y. Sun, X. Hu, W. Luo, H. Xu, C. Hu and Y. Huang, ACS Appl. Mater. Interfaces, 2013, 5, 10145–10150 CrossRef CAS PubMed.
-
(a) W. D. Chemelewski, H.-C. Lee, J.-F. Lin, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 2843–2850 CrossRef CAS PubMed;
(b) J. Jung, K. Song, D. R. Bae, S. W. Lee, G. Lee and Y.-M. Kang, Nanoscale, 2013, 5, 11845–11849 RSC;
(c) Z. Xu, M. Zhang, J. Wu, J. Liang, L. Zhou and B. Lǚ, Water Sci. Technol., 2013, 68, 2178–2185 CrossRef CAS PubMed;
(d) M. Mohapatra and S. Anand, Int. J. Eng. Sci. Technol., 2010, 2, 127–146 Search PubMed.
-
(a) Y. Liu, G.-Q. Qi, C.-L. Liang, R.-Y. Bao, W. Yang, B.-H. Xie and M.-B. Yang, J. Mater. Chem. C, 2014, 2, 3846–38540 RSC;
(b) K. H. Lee, B. Lee, S.-J. Hwang, J.-U. Lee, H. Cheong, O.-S. Kwon, K. Shin and N. H. Hur, Carbon, 2014, 69, 327–335 CrossRef CAS PubMed;
(c) A. T. U. Nugrahenny, J. Kim, S.-K. Kim, D.-H. Peck, S.-H. Yoon and D.-H. Jung, Carbon Lett., 2014, 15, 38–44 CrossRef;
(d) J.-H. Yun, Y. H. Ng, R. J. Wong and R. Amal, ChemCatChem, 2013, 5, 3060–3067 CrossRef CAS PubMed.
-
(a) X. Tian, S. Sarkar, A. Pekker, M. L. Moser, I. Kalinina, E. Bekyarova, M. E. Itkis and R. C. Haddon, Carbon, 2014, 72, 82–88 CrossRef CAS PubMed;
(b) F. T. Thema, M. J. Moloto, E. D. Dikio, N. N. Nyangiwe, L. Kotsedi, M. Maaza and M. Khenfouch, J. Chem., 2013, 150536 Search PubMed.
- A. K. Datta and V. Rakesh, Compr. Rev. Food Sci. Food Saf., 2013, 12, 24–39 CrossRef PubMed.
- C. Qiang, J. Xu, Z. Zhang, L. Tian, S. Xiao, Y. Liu and P. Xu, J. Alloys Compd., 2010, 506, 93–97 CrossRef CAS PubMed.
- P. Klán, J. Literák and S. Relich, J. Photochem. Photobiol., A, 2001, 143, 49–57 Search PubMed.
-
(a) P. Liu and Y. Huang, J. Polym. Res., 2014, 21, 430 Search PubMed;
(b) G. M. Barbosa, M. M. Mosso, C. Vilani, D. R. G. Larrudé, E. C. Romani and L. F. Fernando Jr, Microw. Opt. Technol. Lett., 2014, 56, 560–563 CrossRef PubMed;
(c) J. A. Menéndez, A. Arenillas, B. Fidalgo, Y. Fernández, L. Zubizarreta, E. G. Calvo and J. M. Bermúdez, Fuel Process. Technol., 2010, 91, 1–8 CrossRef PubMed.
-
(a) J. Zheng, H. Lv, X. Lin, G. Ji, X. Li and Y. Du, J. Alloys Compd., 2014, 589, 174–181 CrossRef CAS PubMed;
(b) X. Zhao, Z. Zhang, L. Wang, K. Xi, Q. Cao, D. Wang, Y. Yang and Y. Du, Sci. Rep., 2013, 3, 3421 Search PubMed;
(c) L. Wang, Y. Huang, X. Ding, P. Liu, M. Zong and Y. Wang, Mater. Sci. Eng., B, 2013, 178, 1403–1409 CrossRef CAS PubMed;
(d) L. Wang, Y. Huang, X. Sun, H. Huang, P. Liu, M. Zong and Y. Wang, Nanoscale, 2014, 6, 3157–3164 RSC.
-
(a) Y. L. Liu, X. J. Zhao, X. X. Yang and Y. F. Li, Analyst, 2013, 138, 4526–4531 RSC;
(b) G. Reginald and C. M. Grisham, in Biochemistry, Cengage Learning, Belmont, 4th edn, 2010, part 2, ch. 13, pp. 406–407 Search PubMed;
(c) F. Deyhimi and F. Nami, Int. J. Chem. Kinet., 2012, 44, 699–704 CrossRef CAS PubMed;
(d) L. S. Zamorano, N. F. Cuadrado, P. P. Galende, M. G. Roig and V. L. Shnyrov, J. Biophys. Chem., 2012, 3, 16–28 CAS.
-
(a) Z. Dai, S. Liu, J. Bao and H. Ju, Chem.–Eur. J., 2009, 15, 4321–4326 CrossRef CAS PubMed;
(b) A. K. Dutta, S. K. Maji, D. N. Srivastava, A. Mondal, P. Biswas, P. Paul and B. Adhikary, ACS Appl. Mater. Interfaces, 2012, 4, 1919–1927 CrossRef CAS PubMed.
-
(a) X. Hou, F. Zeng, F. Du and S. Wu, Nanotechnology, 2013, 24, 335502 CrossRef CAS PubMed;
(b) A. Hatamie, B. Zargar and A. Jalali, Talanta, 2014, 121, 234–238 CrossRef CAS PubMed;
(c) A. H. Gore, S. B. Vatre, P. V. Anbhule, S.-H. Han, S. R. Patil and G. B. Kolekar, Analyst, 2013, 138, 1329–1333 RSC;
(d) Z.-X. Wang, C.-L. Zheng, Q.-L. Li and S.-N. Ding, Analyst, 2014, 139, 1751–1755 RSC.
-
(a) J. Zhang, X. Xu and X. Yang, Analyst, 2012, 137, 1556–1558 RSC;
(b) Z.-X. Wang, C.-L. Zheng and S.-N. Ding, RSC Adv., 2014, 4, 9825–9829 RSC;
(c) A. Pandya, K. V. Joshi, N. R. Modi and S. K. Menon, Sens. Actuators, B, 2012, 168, 54–61 CrossRef CAS PubMed;
(d) L.-X. Chen, D.-W. Li, L.-L. Qu, Y.-T. Li and Y.-T. Long, Anal. Methods, 2013, 5, 6579–6582 RSC.
-
(a) X. Cao, W. Lin and L. He, Org. Lett., 2011, 13, 4716–4719 CrossRef CAS PubMed;
(b) L. Zhang, X. Lou, Y. Yu, J. Qin and Z. Li, Macromolecules, 2011, 44, 5186–5193 CrossRef CAS;
(c) C. Kar and G. Das, J. Photochem. Photobiol., A, 2013, 251, 128–133 CrossRef CAS PubMed;
(d) M.-Q. Wang, K. Li, J.-T. Hou, M.-Y. Wu, Z. Huang and X.-Q. Yu, J. Org. Chem., 2012, 77, 8350–8354 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. Fig. S1–S9. See DOI: 10.1039/c4ra05047a |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.