
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halos show the path to perfection: peripheral iodo-substituents improve the efficiencies of bis(diimine)copper(I) dyes in DSCs†
Frederik J.
Malzner
,
Sven Y.
Brauchli
,
Edwin C.
Constable
*,
Catherine E.
Housecroft
* and
Markus
Neuburger
Department of Chemistry, University of Basel, Spitalstrasse 51, CH4056 Basel, Switzerland. E-mail: edwin.constable@unibas.ch; catherine.housecroft@unibas.ch; Tel: +41 61 267 1008
First published on 6th October 2014
Abstract
The homoleptic copper(I) complexes [CuL2][PF6] (L = 4,4′-bis(4-halophenyl)-6,6′-dimethyl-2,2′-bipyridine with halogen = F (2) and Cl (3)) have been prepared and characterized, and their absorption spectroscopic and electrochemical properties compared to that with L = 4,4′-bis(4-bromophenyl)-6,6′-dimethyl-2,2′-bipyridine (4). The synthesis of [CuL2][PF6] (L = 4,4′-bis(4-iodophenyl)-6,6′-dimethyl-2,2′-bipyridine, 5) resulted in a mixture of [Cu(5)2][PF6] and [Cu(5)(MeCN)2][PF6]; variable temperature 1H NMR spectroscopy confirmed that the complexes are in equilibrium in CD3CN solution. The structure of [Cu(5)(MeCN)2][PF6] was determined by single crystal X-ray crystallography, and confirms a distorted tetrahedral geometry for the copper(I) centre. The heteroleptic dyes [Cu(1)(2)]+, [Cu(1)(3)]+, [Cu(1)(4)]+ and [Cu(1)(5)]+ (1 = ((6,6′-dimethyl-[2,2′-bipyridine]-4,4′-diyl)bis(4,1-phenylene))bis(phosphonic acid)) have been assembled by ligand exchange between [CuL2]+ and TiO2 functionalized with the anchoring ligand 1, and the performances of the dyes in fully masked dye-sensitized solar cells (DSCs) have been measured and compared. On the day of DSC fabrication, the trend for the global efficiencies, η, depends on the halo-substituent in the order I > F ≈ Br > Cl. Ripening of the DSCs occurs and after 7 days, the dependence of η on the halo-atom is in the order I > Cl ≈ F ≈ Br; the highest η is 3.16% for [Cu(1)(5)]+ compared to 7.63% for N719. Compared to the other halo-functionalized dyes, [Cu(1)(5)]+ shows an extended spectral response to longer wavelength, with enhanced electron injection. The results of DFT calculations suggest that the better dye performance of [Cu(1)(5)]+ may be associated with improved electron transfer over the halogen of the aryl substituent from the reduced electrolyte. The assembly of anchored dye [Cu(1)(5)]+ by treating functionalized-TiO2 with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of [Cu(MeCN)4]+ and 5, yields a dye which gives a DSC performance that matches that made by ligand exchange using [Cu(5)2][PF6] and [Cu(5)(MeCN)2][PF6].
:1 mixture of [Cu(MeCN)4]+ and 5, yields a dye which gives a DSC performance that matches that made by ligand exchange using [Cu(5)2][PF6] and [Cu(5)(MeCN)2][PF6].
Introduction
Pioneering studies by Sauvage and co-workers in the 1990s established the potential use of copper(I) complexes in dye-sensitized solar cells (DSCs).1 Nonetheless, ruthenium(II)-containing photosensitizers remain the mainstay of conventional Grätzel-type DSCs.2 For sustainable future technologies, it is necessary to develop a materials chemistry in which scarce elements are replaced by more abundant ones. We and others are currently addressing this by focusing attention on the application of copper(I)-containing sensitizers in DSCs.3–14 While the photophysical properties of [Cu(N^N)2]+ (N^N = diimine ligand) are similar to those of ruthenium(II) complexes,15,16 the greater global abundance and lower cost of copper compared to ruthenium make copper-containing dyes relevant and topical. A ground-breaking photoconversion efficiency for a DSC of 4.66% has recently been reported by Odobel, Boujtita and coworkers using a heteroleptic copper(I) dye containing the sterically demanding 6,6′-dimesityl-2,2′-bipyridine-4,4′-dicarboxylic acid anchoring ligand and a 2,2′-bipyridine ancillary ligand with hole-transporting triphenylamino units; the high efficiency of the DSC was achieved in part by using the co-adsorbent chenodeoxycholic acid (cheno).17In contrast to the HETPHEN approach used by the Odobel group,17 we have developed a stepwise method of assembling copper(I) dyes in n-type DSCs commencing with the absorption of an anchoring ligand, Lanchor, onto the n-type semiconductor surface. Subsequent reaction of the functionalized surface with a labile homoleptic complex [Cu(Lancillary)2]+ in solution leads to the formation of surface-bound dye [Cu(Lanchor) (Lancillary)]+.4 Isolation of the heteroleptic complex is, therefore, avoided; indeed, isolation is not usually possible because of rapid equilibration between homo- and heteroleptic cations in solution to give a statistical mixture of species. In an earlier investigation, surface-bound [Cu(Lanchor)(Lancillary)]+ species were characterized by MALDI-TOF mass spectrometry and diffuse reflectance electronic absorption spectroscopy.18 The favoured anchoring ligand is the bis(phosphonic acid) 1 (Scheme 1), with the spacer between the 2,2′-bipyridine and phosphonic acid anchoring domains leading to enhanced performance of the dye.6 Our dye assembly strategy is advantageous in that it permits rapid screening of different families of ancillary ligands, and has recently been implemented by the Robertson group.9 However a disadvantage is the wastage of one equivalent of ancillary ligand, and this is particularly unsatisfactory when synthesis of the latter is a labour intensive multistep procedure.
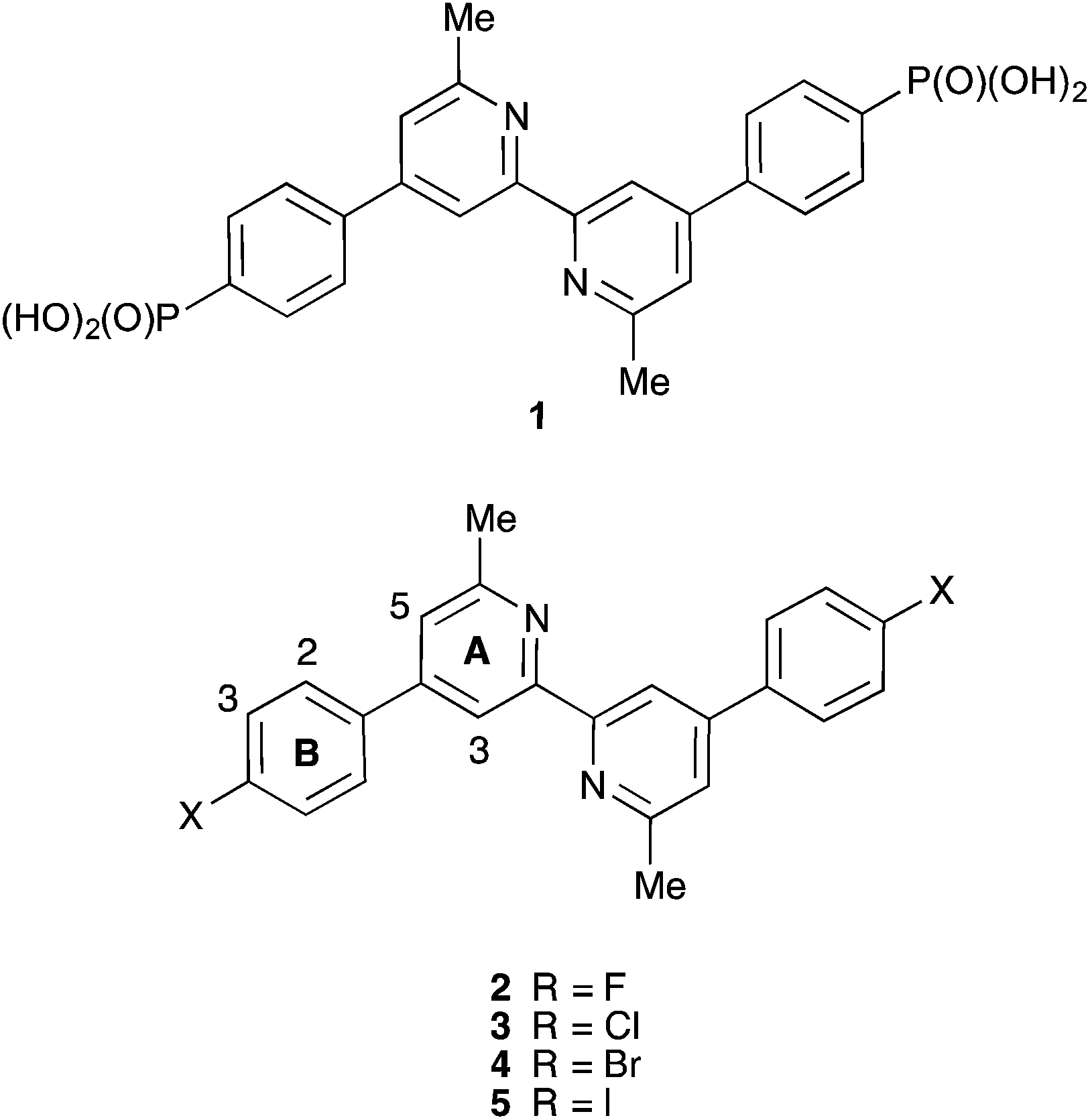 | ||
| Scheme 1 Structures of ligands with atom labelling for NMR spectroscopic assignments. | ||
Recently, we demonstrated that masked DSCs containing the dye [Cu(1)(Lancillary)]+ in which Lancillary is 4,4′-bis(4-bromophenyl)-6,6′-dimethyl-2,2′-bipyridine reached power conversion efficiencies of 2.31%, compared to 8.30% for standard dye N719.6 Since ancillary ligands in n-type dyes typically incorporate electron-donating domains, we were somewhat surprised that dyes containing peripheral bromophenyl substituents (selected to provide an active site for further derivatization) performed relatively well.6 We were, therefore, prompted to study the effects of altering the halo-substituent and now report the remarkably high power conversion efficiencies of DSCs incorporating the series of ancillary ligands 2–5 (Scheme 1). We also illustrate that complete conversion of [Cu(MeCN)4]+ to [Cu(Lancillary)2]+ is not an essential step prior to ligand exchange on the functionalized TiO2 surface, and introduce a stepwise strategy for in situ assembly of the surface-anchored copper(I) dye.
Experimental
General
A Bruker Avance III-500 NMR spectrometer was used to record 1H and 13C NMR spectra, and chemical shifts were referenced to residual solvent peaks with respect to δ(TMS) = 0 ppm. Solution absorption spectra were recorded with a Cary 5000 spectrophotometer and FT-IR spectra of solid samples on a Perkin Elmer UATR Two spectrometer. Electrospray ionization (ESI) mass spectra were recorded on a Bruker esquire 3000plus instrument.Electrochemical measurements were made using a CH Instruments 900B potentiostat with glassy carbon, platinum wire and silver wire as the working, counter, and reference electrodes, respectively. Substrates were dissolved in HPLC grade CH2Cl2 (ca. 10−4 to 10−5 mol dm−3) containing 0.1 mol dm−3 [nBu4N][PF6] as the supporting electrolyte; all solutions were degassed with argon. Cp2Fe was used as internal reference. The scan rate was 0.1 V s−1.
Ground state density functional theory (DFT) calculations were performed using Spartan 14 (v. 1.1.3) at the B3LYP level with a 6-31G* basis set in vacuum. Initial energy optimization was carried out at a semi-empirical (PM3) level.
The external quantum efficiency (EQE) measurements were made using a Spe-Quest quantum efficiency instrument from Rera Systems (Netherlands) equipped with a 100 W halogen lamp (QTH) and a lambda 300 grating monochromator (Lot Oriel). The monochromatic light was modulated to 3 Hz using a chopper wheel (ThorLabs). The cell response was amplified with a large dynamic range IV converter (CVI Melles Griot) and then measured with a SR830 DSP Lock-In amplifier (Stanford Research).
Ligands 16 and 45, [Cu(MeCN)4][PF6]19 and [Cu(4)2][PF6] were prepared by literature methods. Compound 3 was prepared by adapting the literature method,20 replacing 1-(2-oxopropyl)pyridin-1-ium bromide by the corresponding chloride salt. 1-(2-Oxopropyl)pyridin-1-ium chloride was purchased from Fluka.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 146.6 (Ca), 138.3 (CA2), 133.7 (CA1), 130.3 (CA3), 119.8 (Cb), 98.1 (CA4). IR (ν/cm−1): 1670 (s), 1597 (s), 1578 (m), 1553 (m), 1480 (m), 1394 (m), 1312 (w), 1293 (w), 1178 (m), 1057 (w), 981 (s), 802 (s), 726 (s), 584 (s), 515 (w), 453 (m). Found: C 40.57, H 2.48; C18H12I2O2·H2O requires C, 40.63, H, 2.65.
000), 302 (14900). Found: C 74.12, H 4.90, N 7.53; C24H18F2N2·H2O requires C 73.83, H 5.16, N 7.18.
:1) as an off-white solid (93.3 mg, 0.160 mmol, 10.3%). Dec. > 296 °C. 1H NMR (500 MHz, CDCl3) δ/ppm: 8.81 (broadened s, 2H, HA3), 7.87 (m, 4H, HB3), 7.66 (m, 4H, HB2), 7.52 (s, 2H, HA5), 2.90 (s, 6H, HMe). 13C NMR (126 MHz, CDCl3δ/ppm: 158.0 (CA6), 155.3 (CA4), 153.5 (CA2), 138.4 (CB3), 136.2 (CB1), 129.2 (CB3), 122.6 (CA5), 121.9 (CB4), 119.4 (CA3), 23.2 (CMe). IR (ν/cm−1): 1594 (m), 1569 (w), 1542 (w), 1486 (m), 1376 (m), 1004 (s), 817 (s), 745 (s), 511 (w), 472 (s). ESI MS m/z 589.2 [M + H]+ (calc. 589.0). UV-Vis (CH2Cl2, 1.0 × 10−5 mol dm−3) λ/nm 260 (ε/dm3 mol−1 cm−1 46700), sh 309 (23200). Found: C 48.87, H 3.34, N 4.27; C24H18I2N2 requires C 49.00, H 3.08, N 4.76.
200), 321 (39300), 347 (9500), 483 (12600). Found: C 59.91, H 4.30, N 6.75; C48H36CuF10N4P·MeCN requires C 60.39, H 4.08, N 7.04.
500), 322 (46500), 353 sh (10400), 485 (11300). Found: C 53.45, H 3.76, N 5.69; C48H36Cl4CuF6N4P·3H2O requires C 53.72, H 3.94, N 5.22.
O), 146.6 (Ca), 138.3 (CA2), 133.7 (CA1), 130.3 (CA3), 119.8 (Cb), 98.1 (CA4). IR (ν/cm−1): 1670 (s), 1597 (s), 1578 (m), 1553 (m), 1480 (m), 1394 (m), 1312 (w), 1293 (w), 1178 (m), 1057 (w), 981 (s), 802 (s), 726 (s), 584 (s), 515 (w), 453 (m). Found: C 40.57, H 2.48; C18H12I2O2·H2O requires C, 40.63, H, 2.65.
000), 302 (14900). Found: C 74.12, H 4.90, N 7.53; C24H18F2N2·H2O requires C 73.83, H 5.16, N 7.18.
:1) as an off-white solid (93.3 mg, 0.160 mmol, 10.3%). Dec. > 296 °C. 1H NMR (500 MHz, CDCl3) δ/ppm: 8.81 (broadened s, 2H, HA3), 7.87 (m, 4H, HB3), 7.66 (m, 4H, HB2), 7.52 (s, 2H, HA5), 2.90 (s, 6H, HMe). 13C NMR (126 MHz, CDCl3δ/ppm: 158.0 (CA6), 155.3 (CA4), 153.5 (CA2), 138.4 (CB3), 136.2 (CB1), 129.2 (CB3), 122.6 (CA5), 121.9 (CB4), 119.4 (CA3), 23.2 (CMe). IR (ν/cm−1): 1594 (m), 1569 (w), 1542 (w), 1486 (m), 1376 (m), 1004 (s), 817 (s), 745 (s), 511 (w), 472 (s). ESI MS m/z 589.2 [M + H]+ (calc. 589.0). UV-Vis (CH2Cl2, 1.0 × 10−5 mol dm−3) λ/nm 260 (ε/dm3 mol−1 cm−1 46700), sh 309 (23200). Found: C 48.87, H 3.34, N 4.27; C24H18I2N2 requires C 49.00, H 3.08, N 4.76.
200), 321 (39300), 347 (9500), 483 (12600). Found: C 59.91, H 4.30, N 6.75; C48H36CuF10N4P·MeCN requires C 60.39, H 4.08, N 7.04.
500), 322 (46500), 353 sh (10400), 485 (11300). Found: C 53.45, H 3.76, N 5.69; C48H36Cl4CuF6N4P·3H2O requires C 53.72, H 3.94, N 5.22.
Crystallography
Single crystal data were collected on a Bruker APEX-II diffractometer with data reduction, solution and refinement using the programs APEX22 and CRYSTALS.23 The ORTEP-type diagram and structure analysis used Mercury v. 3.0.24,25350 reflections, 5520 unique, Rint = 0.027. Refinement of 5460 reflections (380 parameters) with I > 3σ(I) converged at final R1 = 0.0250 (R1 all data = 0.0251), wR2 = 0.0261 (wR2 all data = 0.0263), gof = 1.0853.
DSC fabrication and measurements
DSCs were prepared modifying the method of Grätzel.26,27 Commercial Solaronix Test Cell Titania Electrodes were used. The electrodes were rinsed with EtOH and sintered at 450 °C for 30 min, then cooled to ≈80 °C and immersed in a 1 mM DMSO solution of the anchoring ligand 1 for 24 h. The colourless electrode was removed from the solution, washed with DMSO and EtOH and dried at ≈60 °C (heat gun). The electrode with adsorbed 1 was immersed in a 0.1 mM CH2Cl2 solution of each copper(I) complex for ≈68 h. For the final set of DSCs (see text), the electrode with adsorbed anchoring ligand was immersed in a CH2Cl2 solution containing equimolar (0.1 mM) amounts of [Cu(MeCN)4][PF6] and ancillary ligand 5 for 68 h; during this time, the electrodes turned pale red-orange.Each reference electrode was prepared by dipping a Solaronix Test Cell Titania Electrode in a 0.3 mM EtOH solution of standard dye N719 (Solaronix) for ≈68 h. The electrodes were washed with the same solvent used in the dipping period and dried at ≈60 °C (heat gun). A Solaronix Test Cell Platinum Electrode was used for the counter electrode, and it was heated for 30 min at 450 °C (heating plate) to remove impurities.
The two electrodes were combined using thermoplast hot-melt sealing foil (Solaronix Test Cell Gaskets) by heating while pressing them together. The electrolyte was a mixture of LiI (0.1 mol dm−3), I2 (0.05 mol dm−3), 1-methylbenzimidazole (0.5 mol dm−3) and 1-butyl-3-methylimidazolinium iodide (0.6 mol dm−3) in 3-methoxypropionitrile; it was introduced into the DSC by vacuum backfilling. The hole on the counter electrode was sealed using hot-melt sealing foil (Solaronix Test Cell Sealings) and a cover glass (Solaronix Test Cell Caps). Measurements were made by irradiating from behind using a light source SolarSim 150 (100 mW cm−2 = 1 sun). The power of the simulated light was calibrated by using a reference Si cell.
Results and discussion
Ligand synthesis and characterization
We have previously used the strategy of Kröhnke28 to prepare compounds 320 and 45 and now extend the series of halo-derivatives to compounds 2 and 5 (Scheme 2). The yield of 2 was only moderate (52.8%), and 5 proved very difficult to purify and pure material was obtained in only 10.3% yield. The electrospray mass spectra of 2 and 5 exhibited base peaks at m/z 373.3 and 589.2, respectively, corresponding to [M + H]+. The 1H and 13C NMR spectra were assigned by COSY, HMQC and HMBC methods and are consistent with the disubstitution pattern shown in Scheme 2. Broadening of the signals for HA3 and HA5 (see Scheme 1 for atom labelling) is most likely associated with rotation of the 4-halophenyl groups on the NMR timescale. At 500 MHz, values of FWHM for the signals for HA3 and HA5 are 36 and 12 Hz, respectively in 2, and 21 and 17 Hz in 5. The solution absorption spectra of compounds 2–5 are compared in Fig. 1, the intense high energy bands arising from π* ← π and π* ← n transitions. The highest energy absorption shifts from 248 nm in 2 to 260 nm in 5.
 | ||
| Scheme 2 Synthesis of ligands 2 and 5 using the Kröhnke approach. Atom numbering for NMR assignments of the precursors. | ||
 | ||
| Fig. 1 Absorption spectra of CH2Cl2 solutions of ligands 2–5 (1.0 × 10−5 mol dm−3). | ||
Synthesis and characterization of copper(I) complexes
We have described the complex [Cu(4)2][PF6] previously,6 and the same simple strategy was adopted to prepare analogous homoleptic complexes containing ligands 2, 3 or 5. Treatment of [Cu(MeCN)4][PF6] with two equivalents of 2 or 3 gave [Cu(2)2][PF6] or [Cu(3)2][PF6] as orange-red solids in 71.7 and 95.2% yields. The highest mass peak envelope in the electrospray mass spectrum of each complex corresponded to the [M − PF6]+ ion, and isotope patterns matched those calculated. The base peak in each spectrum arose from [L + H]+ (L = 2 or 3).The 1H and 13C NMR spectra of a CD3CN solution of [Cu(2)2][PF6] were consistent with a single ligand environment, and the aromatic region of the 1H NMR spectrum is shown in Fig. 2a. Coupling to 19F gives characteristic signals for protons HB2 and HB3 and doublets for all ring B resonances in the 13C NMR spectrum (see Scheme 1 for numbering), and the assignments were confirmed through the HMBC and HMQC spectra. The methyl groups give rise to sharp singlets at δ 2.35 and 25.2 ppm in the 1H and 13C{1H} NMR spectra, respectively, of [Cu(2)2][PF6], and these shifts are unchanged on going to [Cu(3)2][PF6]. The aromatic region of the 1H NMR spectrum of a CD3CN solution of [Cu(3)2][PF6] is shown in Fig. 2b. The change from fluoro to chloro substituent has the greatest effect on HB3 in keeping with expectations.29
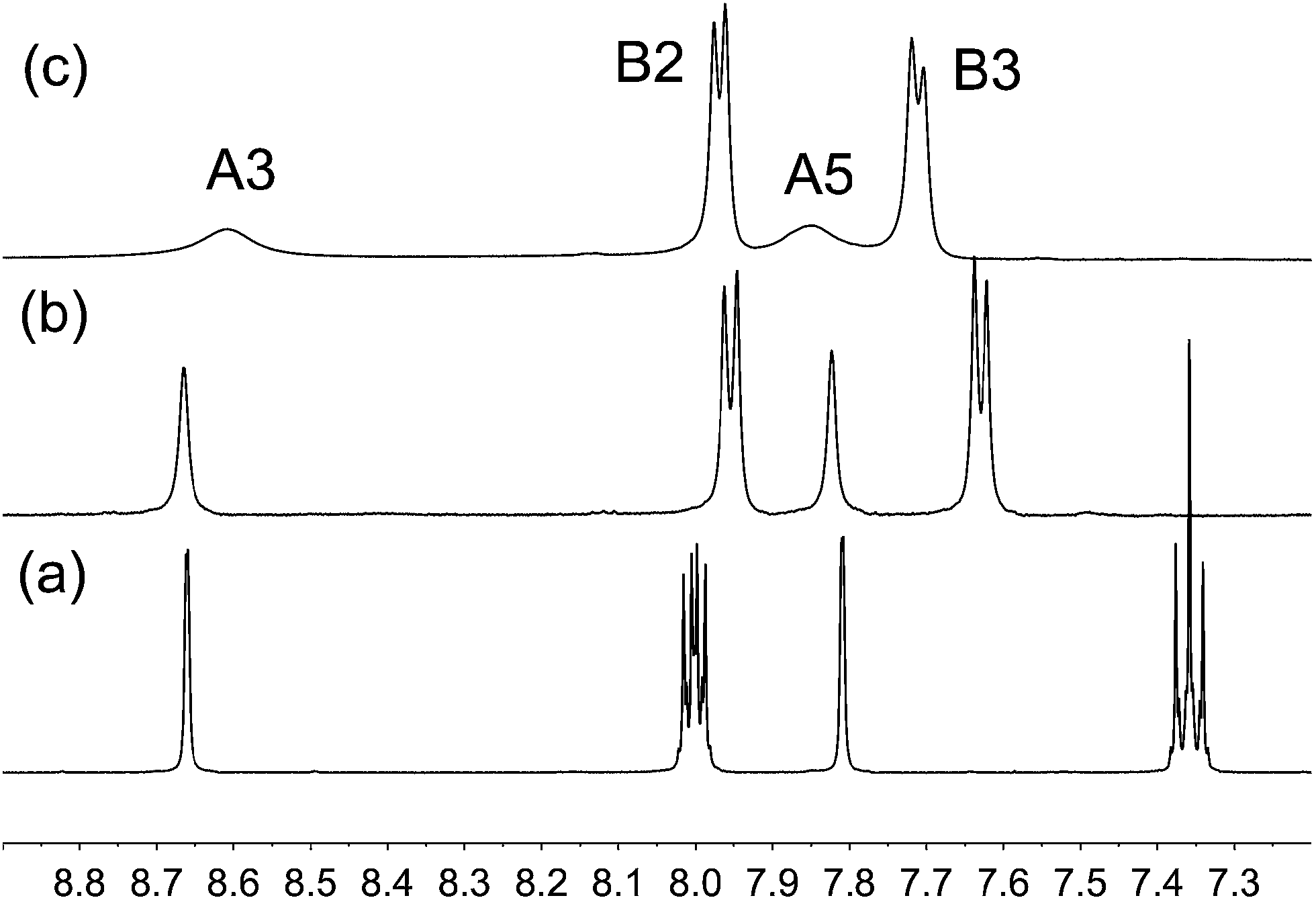 | ||
| Fig. 2 500 MHz 1H NMR spectra (aromatic region only) of CD3CN solutions of (a) [Cu(2)2][PF6], (b) [Cu(3)2][PF6] and (c) [Cu(5)2][PF6] (see text). | ||
Preparation of the iodo-containing complex [Cu(5)2][PF6] proved more problematical. The method used was as for the fluoro-, chloro- and bromo-containing complexes, but repeated purification of the bulk sample failed to produce analytically pure [Cu(5)2][PF6]. In contrast to the sharp singlet for the methyl protons in [Cu(2)2][PF6] and [Cu(3)2][PF6], the room temperature 1H NMR spectrum of the iodo-containing product showed a very broad signal centred at δ 2.52 ppm with FWHM ∼145 Hz. The 13C NMR resonance for CMe could not be resolved at 295 K and no cross peak for the HMe signal was observed in the HMQC spectrum. The signals for HA3 and HA5 were also broad (Fig. 2c). Cooling the sample to 240 K led to collapse of the broad peaks and appearance of two sets of signals (Fig. 3), consistent with two environments for coordinated ligand 5 (the free ligand is poorly soluble in acetonitrile and therefore signals arising from ligand can be discounted). The separations of pairs of signals for HMe indicate that the two methyl groups are significantly different in the two species (δ 2.26 and 2.83 ppm), and similarly for the two HA3 protons (δ 8.42 and 8.66 ppm). The local environment of HA5 appears similar in the two species, and similarly for HB2 and HB3. We propose that the bulk material contains a mixture of [Cu(5)2][PF6] and [Cu(5)(MeCN)2][PF6], and that at 295 K, ligand exchange occurs between [Cu(5)(MeCN)2]+ and [Cu(5)2]+. This is supported by the electrospray mass spectrum of the bulk material which exhibited peak envelopes with a characteristic copper isotope pattern at m/z 1239.3 and 692.2 arising from [Cu(5)2]+ and [Cu(5)(MeCN)]+; the base peak (m/z 589.2) was assigned to [5 + H]+.
 | ||
| Fig. 3 Variable temperature 500 MHz 1H NMR spectra of the bulk material containing [Cu(5)2][PF6] and [Cu(5)(MeCN)2][PF6]. * = residual H2O in CD3CN. | ||
The identity of [Cu(5)(MeCN)2][PF6] as one component of the mixture was confirmed by a single crystal structure determination of orange blocks that grew during recrystallization of the bulk material (see Experimental section). Fig. 4 shows the structure of the [Cu(5)(MeCN)2]+ cation in [Cu(5)(MeCN)2][PF6]. The complex crystallizes in the monoclinic space group C2/c. Atom Cu1 is in a distorted tetrahedral environment; the bite angle of the bpy unit is 81.06(7)°, and other N–Cu–N bond angles range from 106.24(9) to 124.31(8)° (caption to Fig. 4). The bpy domain deviates from planarity with the angle between the planes of the pyridine rings being 13.8°. The phenyl ring containing C12 is twisted through 15.5° with respect to the plane of the pyridine ring with N1, while the corresponding angle for the rings containing C18 and N2 is 38.7°. The difference in twist angles is associated with the packing of the cations. Face-to-face π-stacking occurs between phenylbpy domains involving the phenyl ring containing atom C12 and this leads to infinite columns of cations running parallel to the c-axis (Fig. 5a). The stacks are built up by alternating operations of a 2-fold axis followed by an inversion centre. The asymmetric unit contains two half-anions; each [PF6]− is ordered, and atoms P1 and P2 are each located on a 2-fold axis. Cation⋯anion interactions involve extensive CH⋯F contacts. For the anion containing P1 (green in Fig. 5b), these contacts involve arene CH units; in addition this anion exhibits a short F⋯I contact (F3⋯I2i = 3.406(1) Å, symmetry code i = 1/2 − x, −1/2 + y, 1/2 − z). In contrast, the anion containing P2 (red in Fig. 5b) sits between the π-stacked cations and interacts with methyl groups of both coordinated ligands 5 and MeCN.
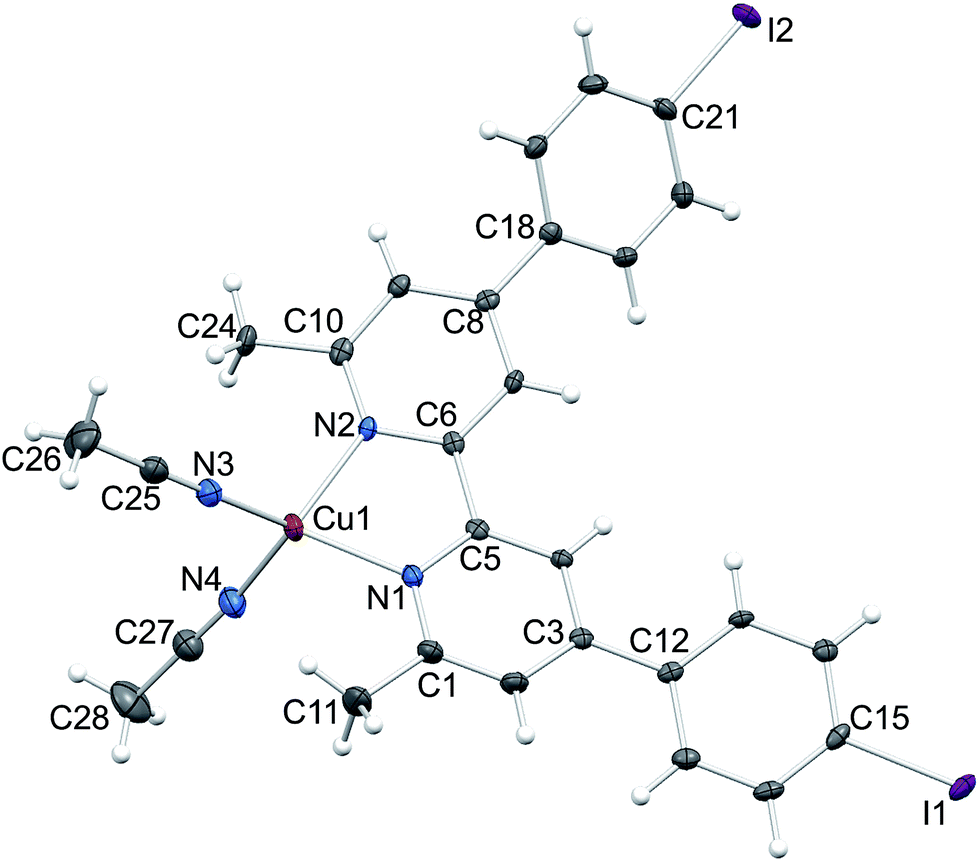 | ||
| Fig. 4 Structure of the [Cu(5)(MeCN)2]+ cation in [Cu(5)(MeCN)2][PF6] with ellipsoids plotted at 50% probability level. Selected bond parameters: Cu1–N1 = 2.0515(19), Cu1–N2 = 2.0527(18), Cu1–N3. 1.949(2), Cu1–N4 = 1.968(2), I1–C15 = 2.092(2), I2–C21 = 2.098(2) Å; N1–Cu1–N2 = 81.06(7), N1–Cu1–N3 = 124.31(8), N2–Cu1–N3 = 114.59(8), N1–Cu1–N4 = 110.66(8), N2–Cu1–N4 = 119.27(8), N3–Cu1–N4 = 106.24(9)°. | ||
 | ||
| Fig. 5 (a) Face-to-face π-stacking of phenylbpy domains (space-filling representation) involving the phenyl ring with C12 leading to infinite assemblies following the c-axis. View down the c-axis showing the two different [PF6]− environments. Anion with P1 is shown in green, and with P2, in red; CH⋯F contacts are shown by red hashed lines. | ||
Solution absorption spectra and electrochemistry
Absorption spectroscopic and electrochemical data were recorded only for [Cu(2)2][PF6] and [Cu(3)2][PF6] since the homoleptic complex with ligand 5 could not be obtained free of [Cu(5)(MeCN)2][PF6]. The absorption spectra are shown in Fig. 6 and are compared with that of [Cu(4)2][PF6].6 Approximate doubling of the extinction coefficients on going from the free ligands (Fig. 1) to the complexes is consistent with the formation of the homoleptic species. The intense, high-energy bands are similar for all the complexes, and a small red-shift is observed for the MLCT band from 483 nm for the fluoro-containing complex to 488 nm6 for the bromo-derivative. The mixture of [Cu(5)2][PF6] and [Cu(5)(MeCN)2][PF6] has an MLCT maximum at 488 nm.
 | ||
| Fig. 6 Solution (CH2Cl2) absorption spectra of the fluoro, chloro and bromo-containing complexes [Cu(2)2][PF6], [Cu(3)2][PF6] and [Cu(4)2][PF6]. | ||
The electrochemical behaviour of [Cu(2)2][PF6], [Cu(3)2][PF6] and [Cu(4)2][PF6] are compared in Table 1, and a representative cyclic voltammogram (with internal Fc/Fc+ reference) is shown in Fig. 7. Each complex exhibits a copper-centred oxidation process and the introduction of the F, Cl or Br substituents has only a small effect on its potential. In each of [Cu(2)2][PF6] and [Cu(3)2][PF6], the irreversible ligand-centred reduction process ca. −2.0 V is more pronounced in the first cycle than in subsequent scans.
| Complex | E ox1/2/V (Epc − Epa/mV) | E red1/2/V | E red1/2/V | Ref. |
|---|---|---|---|---|
| [Cu(2)2][PF6] | +0.39 (74) | −2.08ir | −2.42ir | this work |
| [Cu(3)2][PF6] | +0.43 (67) | −2.06ir | −2.42ir | this work |
| [Cu(4)2][PF6] | +0.42 (94) | −2.16ir | 6 |
 | ||
| Fig. 7 Cyclic voltammogram for a CH2Cl2 solution of [Cu(2)2][PF6]; scan rate 0.1 V s−1 and referenced internally to Fc/Fc+. | ||
Comparison of the performances of the copper(I) dyes in DSCs
Surface-immobilized copper(I) dyes were assembled on commercial titania electrodes by initial adsorption of the phosphonic acid anchoring ligand 1 followed by treatment with CH2Cl2 solutions of the labile30–32 homoleptic complexes [CuL2][PF6] (L = 2, 3 or 4) or a mixture of [Cu(5)2][PF6] and [Cu(5) (MeCN)2][PF6] (Scheme 3). All cells were fully masked,33 and were prepared in duplicate. The electrolyte contained the I3−/I− redox shuttle. The DSC parameters measured for each cell are given in Table 2 and are compared to data for a DSC with standard dye N719. The right-hand column in Table 2 gives the energy conversion efficiencies, η, relative to N719 arbitrarily set to 100%. We have recently adopted this presentation of results to provide a valid means of comparing data recorded using different solar simulators.34 For each DSC, the efficiencies were measured on the day of sealing the cell and then three and seven days later. | ||
| Scheme 3 Assembly of heteroleptic copper(I) dyes using anchoring ligand 1. | ||
| Dye | J SC/mA cm−2 | V OC/mV | ff | η/% | Relative η/% |
|---|---|---|---|---|---|
| a N719 reference cell used in conjunction with DSCs containing dyes [Cu(1)(2)]+ and [Cu(1)(3)]+. b N719 reference cell used with dyes [Cu(1)(4)]+ and [Cu(1)(5)]+. | |||||
| On the day of sealing the cell | |||||
| [Cu(1)(2)]+ | 6.70 | 527 | 68.3 | 2.41 | 33.8 |
| [Cu(1)(2)]+ | 6.37 | 544 | 70.0 | 2.42 | 33.9 |
| [Cu(1)(3)]+ | 5.59 | 519 | 71.5 | 2.08 | 29.2 |
| [Cu(1)(3)]+ | 6.00 | 522 | 69.5 | 2.18 | 30.6 |
| N719a | 16.66 | 637 | 67.2 | 7.13 | 100 |
| [Cu(1)(4)]+ | 6.64 | 514 | 69.3 | 2.37 | 31.9 |
| [Cu(1)(4)]+ | 6.49 | 525 | 71.7 | 2.45 | 33.0 |
| [Cu(1)(5)]+ | 6.68 | 582 | 74.0 | 2.88 | 38.8 |
| [Cu(1)(5)]+ | 6.92 | 576 | 72.6 | 2.89 | 38.9 |
| N719b | 16.08 | 641 | 72.0 | 7.43 | 100 |
|
|||||
| 3 days after sealing the cell | |||||
| [Cu(1)(2)]+ | 6.49 | 544 | 69.8 | 2.46 | 32.4 |
| [Cu(1)(2)]+ | 6.19 | 570 | 69.5 | 2.45 | 32.2 |
| [Cu(1)(3)]+ | 6.28 | 566 | 69.7 | 2.48 | 32.7 |
| [Cu(1)(3)]+ | 6.69 | 562 | 68.0 | 2.56 | 33.7 |
| N719a | 16.82 | 667 | 67.7 | 7.59 | 100 |
| [Cu(1)(4)]+ | 5.85 | 554 | 68.1 | 2.21 | 29.0 |
| [Cu(1)(4)]+ | 6.18 | 562 | 70.3 | 2.44 | 32.1 |
| [Cu(1)(5)]+ | 6.81 | 604 | 74.0 | 3.04 | 39.9 |
| [Cu(1)(5)]+ | 7.07 | 586 | 72.6 | 3.01 | 39.6 |
| N719b | 15.87 | 664 | 72.3 | 7.61 | 100 |
|
|||||
| 7 days after sealing the cell | |||||
| [Cu(1)(2)]+ | 6.62 | 556 | 69.1 | 2.54 | 33.7 |
| [Cu(1)(2)]+ | 6.20 | 578 | 68.9 | 2.47 | 32.8 |
| [Cu(1)(3)]+ | 6.27 | 559 | 68.1 | 2.39 | 31.7 |
| [Cu(1)(3)]+ | 6.95 | 565 | 66.2 | 2.60 | 34.5 |
| N719a | 16.42 | 692 | 66.3 | 7.53 | 100 |
| [Cu(1)(4)]+ | 6.55 | 559 | 61.6 | 2.26 | 29.6 |
| [Cu(1)(4)]+ | 6.46 | 561 | 67.5 | 2.45 | 32.1 |
| [Cu(1)(5)]+ | 7.10 | 604 | 73.6 | 3.16 | 41.4 |
| [Cu(1)(5)]+ | 7.42 | 579 | 71.5 | 3.07 | 40.2 |
| N719b | 16.21 | 662 | 71.1 | 7.63 | 100 |
The first point to note is that the DSC parameters for the dye [Cu(1)(4)]+ (bromo-substituents) are comparable with those we have previously reported,6 despite differences in electrode origins. In the present work, the photoanodes are commercially available titania electrodes which include a scattering layer; in our previous study, screen-printed electrodes with scattering layer were prepared in our laboratory. A second important point is that the data in Table 2 for corresponding pairs of DSCs confirm reproducibility of measurements.
On the day of cell fabrication, all four dyes (Table 2) perform relatively well with the global efficiencies, η, dependent upon the halo-substituent in the order I > F ≈ Br > Cl. Upon aging of the DSCs, there is a general trend for improvement of performance (Table 2). Over a three day period, the DSCs containing the iodo-substituted dye [Cu(1)(5)]+ show a ripening effect with η increasing from 2.88 to 3.01%, and 2.89 to 3.04% for the two independent DSCs. After a further four days, the efficiencies increase to 3.16 and 3.07%, respectively. This enhancement is likely associated with initial dye aggregation on the surface followed by molecular reorganization over time.35–37 In terms of η, the best performing DSCs after 7 days are those with iodo-dye [Cu(1)(5)]+ which show power conversion efficiencies of 40.2 or 41.4% with respect to N719 set at 100%. For the aged DSCs, the dependence of η on the halo-substituent follows the trend I > Cl ≈ F ≈ Br.
Fig. 8 shows J–V curves for DSCs containing anchored dyes [Cu(1)(2)]+, [Cu(1)(3)]+ and [Cu(1)(5)]+; data for the bromo-containing dye [Cu(1)(4)]+ essentially replicate those already published.6 All J–V curves show good fill factors. The most significant features in Fig. 8 are the enhancements in both short-circuit current density and open-circuit voltage over time for the chloro- and iodo-containing dyes (Fig. 8b and c). For [Cu(1)(5)]+ (iodo), a maximum value of VOC = 604 mV is achieved after 3 days with no further improvement over the next 4 days (Fig. 8c); 604 mV compares with VOC(max) = 664 mV for N719 measured under the same conditions are the copper(I) dyes (Table 2). Enhancement in the open-circuit voltage with aging of the DSC has also been noted for other copper(I) dyes.38
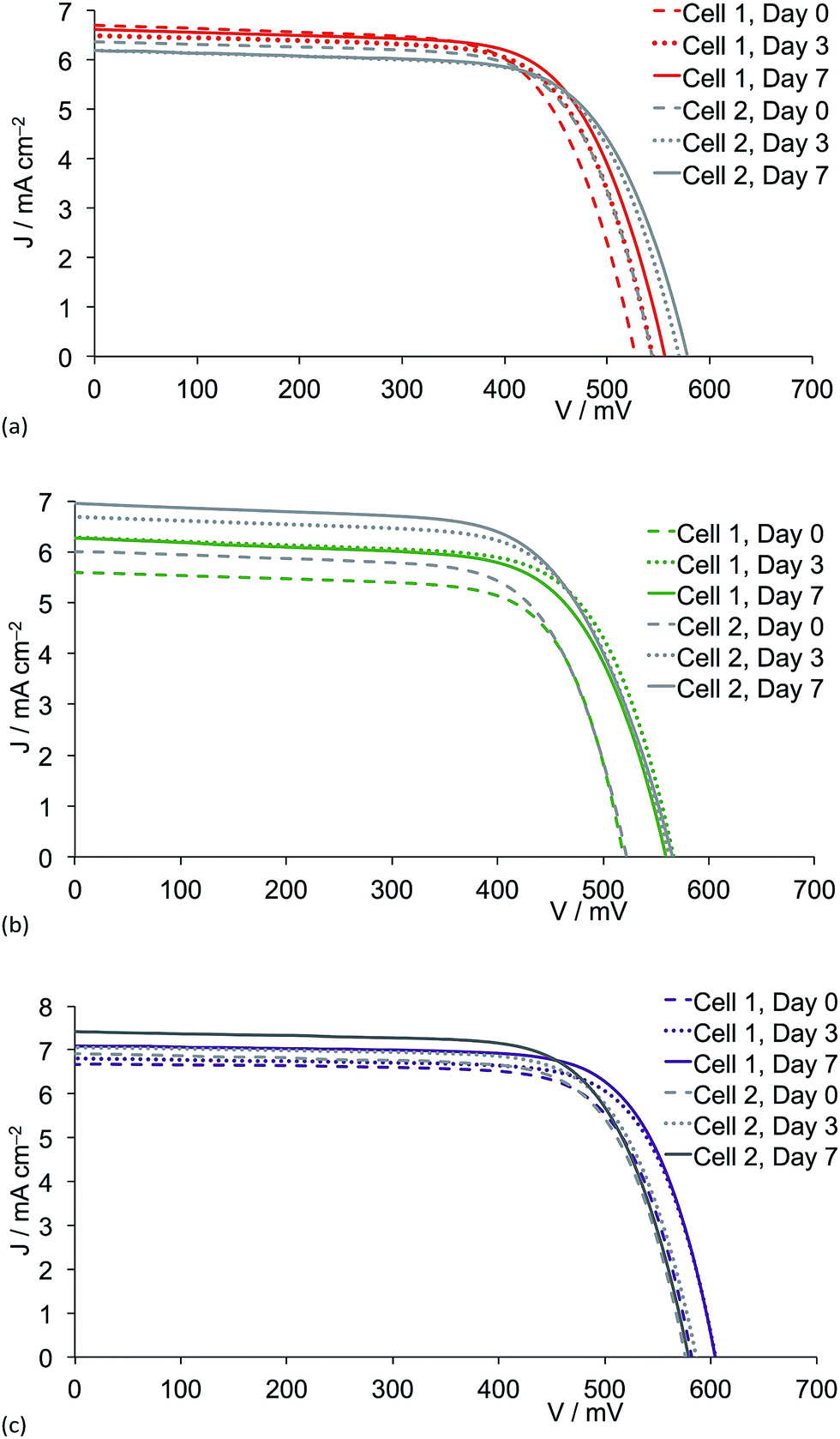 | ||
| Fig. 8 J–V curves for two independent DSCs containing dye (a) [Cu(1)(2)]+ (fluoro substituents), (b) [Cu(1)(3)]+ (chloro substituents) and (c) [Cu(1)(5)]+ (iodo substituents) measured over a 7 day period after sealing the cells. | ||
The EQE spectra of the DSCs were measured over a period of a week following cell fabrication. All show EQE maxima corresponding to λmax in the range 460–480 nm (Table 3). The values of EQEmax for the copper(I) dyes compare to EQEmax ≈ 75% for N719 (Fig. 9). Within experimental error, the EQE values for the cells containing [Cu(1)(2)]+ and [Cu(1)(4)]+ do not change with time, remaining around 46–47%. For DSCs with [Cu(1)(3)]+ (chloro) or [Cu(1)(5)]+ (iodo), a small enhancement in the EQE is observed up to 50 or 51%, respectively after 3 days with no further improvement (Fig. 9). Fig. 10 compares the EQE spectra for DSCs containing [Cu(1)(2)]+, [Cu(1)(3)]+, [Cu(1)(4)]+ or [Cu(1)(5)]+. The most significant feature is the extended spectral response to longer wavelength for [Cu(1)(5)]+ compared to the other halo-functionalized dyes. This indicates improved electron injection and is consistent with the higher JSC values observed for DSCs containing [Cu(1)(5)]+ (Table 2 and Fig. 8). The differences in the EQE spectra in Fig. 10 also correspond to the dependence of η on the halo-substituent (I > Cl ≈ F ≈ Br) for the aged cells described earlier in this section.
| Anchored dye | Day 0 | Day 3 | ||
|---|---|---|---|---|
| λ max/nm | EQEmax/% | λ max/nm | EQEmax/% | |
| [Cu(1)(2)]+ | 480 | 46.1 | 470 | 46.6 |
| [Cu(1)(2)]+ | 480 | 46.4 | 470 | 46.0 |
| [Cu(1)(3)]+ | 480 | 44.1 | 470 | 46.9 |
| [Cu(1)(3)]+ | 470 | 47.8 | 460 | 50.2 |
| [Cu(1)(4)]+ | 470 | 48.3 | 460 | 46.2 |
| [Cu(1)(4)]+ | 470 | 48.1 | 470 | 47.9 |
| [Cu(1)(5)]+ | 470 | 48.0 | 470 | 49.4 |
| [Cu(1)(5)]+ | 470 | 48.2 | 470 | 51.4 |
 | ||
| Fig. 9 EQE spectra for duplicate DSCs functionalized with [Cu(1)(5)]+ measured on the day of sealing the cell (day 0) and 3 and 7 days later, compared to the EQE spectrum of a DSC with N719. | ||
 | ||
| Fig. 10 Comparison of the EQE spectra for DSCs containing dyes [Cu(1)(2)]+ (fluoro), [Cu(1)(3)]+ (chloro), [Cu(1)(4)]+ (bromo) and [Cu(1)(5)]+ (iodo) measured 3 days after sealing the cells. | ||
In situ dye assembly
Because the assembly of the best-performing dye [Cu(1)(5)]+ was carried out using a mixture of [Cu(5)2]+ and [Cu(5)(MeCN)2]+ in the dipping process, we decided to assess a new strategy which eliminates the need to prepare the homoleptic copper(I) complex. The electrode functionalized with anchoring ligand 1 was immersed in a CH2Cl2 solution containing a 1:1 mixture of [Cu(MeCN)4][PF6] and 5 (Scheme 4); the concentration of each was 0.1 mM. Over the dipping period of 68 hours, the electrode became pale red-orange and the colour persisted after drying. Duplicate cells were prepared and the DSC characteristics compared to an N719 standard are given in Table 4.
 | ||
| Scheme 4 Stepwise assembly of titania-anchored [Cu(1)(5)]+. | ||
| Dye | J SC/mA cm−2 | V OC/mV | ff | η/% | Relative η/% |
|---|---|---|---|---|---|
| On the day of sealing the cell | |||||
| [Cu(1)(5)]+ | 6.82 | 551 | 69.1 | 2.59 | 34.4 |
| [Cu(1)(5)]+ | 7.01 | 559 | 70.2 | 2.75 | 36.5 |
| N719 | 16.88 | 641 | 69.6 | 7.54 | 100 |
|
|||||
| 3 days after sealing the cell | |||||
| [Cu(1)(5)]+ | 6.97 | 584 | 69.6 | 2.83 | 37.3 |
| [Cu(1)(5)]+ | 6.93 | 588 | 67.6 | 2.75 | 36.3 |
| N719 | 16.54 | 671 | 68.2 | 7.58 | 100 |
|
|||||
| 7 days after sealing the cell | |||||
| [Cu(1)(5)]+ | 6.85 | 587 | 69.7 | 2.80 | 36.0 |
| [Cu(1)(5)]+ | 6.96 | 587 | 66.3 | 2.71 | 34.9 |
| N719 | 16.55 | 684 | 68.7 | 7.77 | 100 |
The performances of the duplicate DSCs are similar and exhibit efficiencies of 2.80 or 2.71% after 7 days. One cell shows an enhanced performance over the 7 days after sealing the DSCs (Table 4) and both cells exhibit improved VOC but little change in JSC (Fig. 11). The EQE spectra for the two DSCs show maxima at 46.0 and 46.1% (λmax = 470 nm) on the day of cell fabrication and these values vary little over a 7 day period (Fig. 12). Overall, the performances of the DSCs containing dye [Cu(1)(5)]+ assembled in situ are comparable with those of the DSCs made by ligands exchange. The differences in performance (compare the entries for [Cu(1)(5)]+ in Tables 2 and 4) are not significant and imply that isolation of the homoleptic copper(I) complex is not an essential part of the cell-assembly process.
 | ||
| Fig. 11 J–V curves measured over a 7 day period for one of the DSCs containing [Cu(1)(5)]+ formed in situ by stepwise assembly. | ||
 | ||
| Fig. 12 EQE spectra measured over a 7 day period for one of the DSCs containing [Cu(1)(5)]+ formed in situ by stepwise assembly. | ||
HOMO and LUMO characteristics
We have used ground state DFT calculations to gain some insight into the origin of the surprisingly good performance of dyes containing iodo-substituted ligand 5. We have previously shown that the choice of atomic orbital basis set (6-311++G** basis set on all atoms, 6-311++G** on copper and 6-31G* basis set on C, H and N, or 6-31G* basis set on all atoms) has a strong influence on the calculated absorption spectra of representative bis(diimine) copper(I) dyes, while the orbital characteristics of HOMOs and LUMOs are essentially unaffected.39 Thus, for qualitative assessment of the MOs of [Cu(1)(2)]+, [Cu(1)(3)]+, [Cu(1)(4)]+ and [Cu(1)(5)]+, we opted to use a 6-31G* basis set on all atoms for reasons of computational efficiency.DFT calculations on the ground state, energy minimized structures of [Cu(1)(2)]+, [Cu(1)(3)]+, [Cu(1)(4)]+ and [Cu(1)(5)]+ showed that the energies and characteristics of the LUMO and LUMO+1 of the four complexes are similar. The LUMO is centred on the anchoring ligand while the LUMO+1 is principally localized on the bpy domain of the ancillary ligand. These MOS are shown for the iodo-complex in Fig. 13a and b. The close similarity in the orbital characteristics for the four dyes suggests that the enhanced performance of [Cu(1)(5)]+ is not associated with a tuning of the properties of the lowest lying vacant MOs leading to improved electron injection.
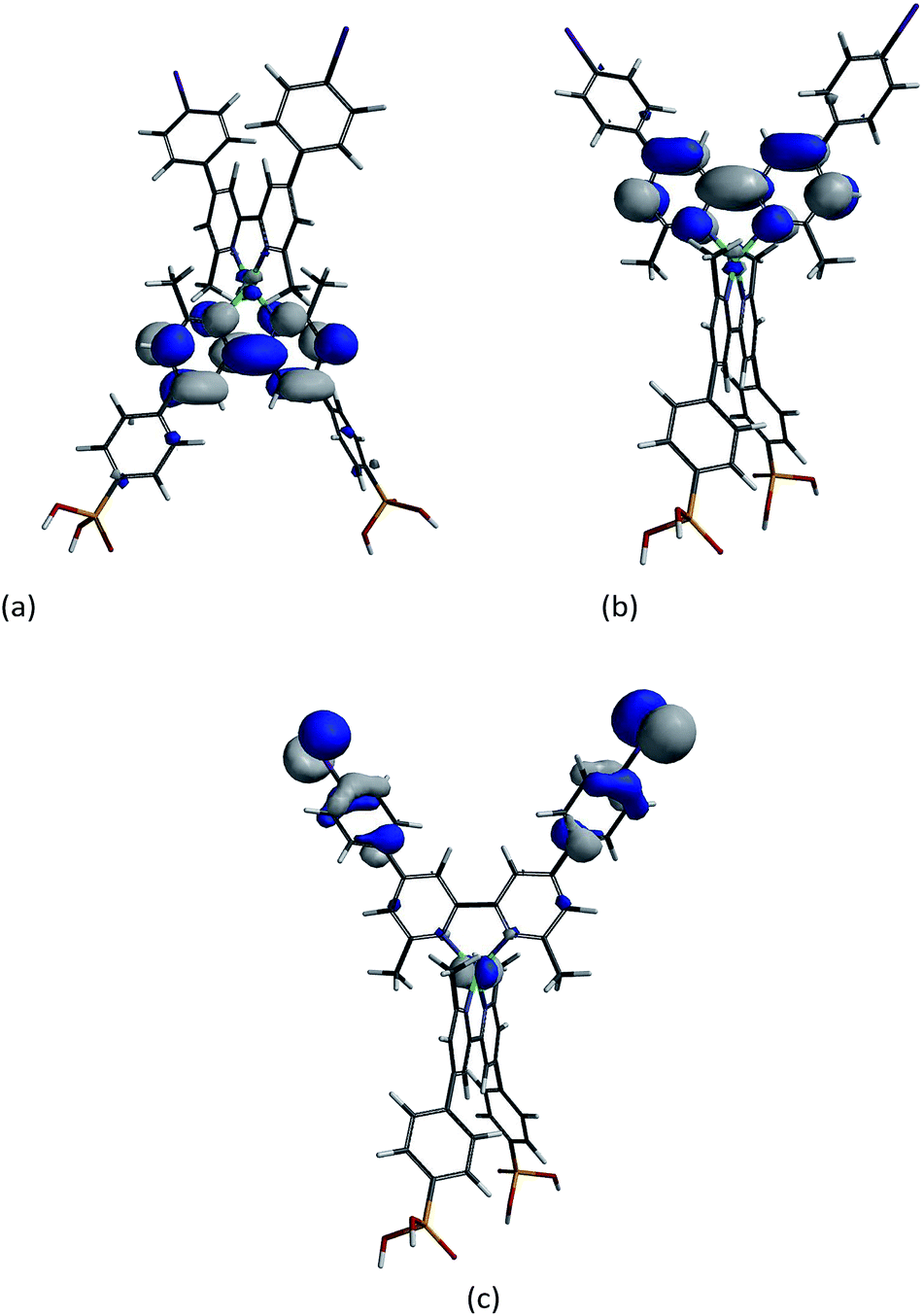 | ||
| Fig. 13 (a) LUMO, (b) LUMO+1, and (c) HOMO−3 of [Cu(1)(5)]+ (ligand 5 is at the top of each diagram). | ||
The DFT calculations indicate that the HOMO of each ground-state dye is mainly based on copper, as are the next two highest occupied MOs. Ancillary ligand character is present in HOMO−3 and HOMO−4 in [Cu(1)(5)]+, with a dominant contribution from the iodophenyl substituent (Fig. 13c). Significantly, the corresponding contributions by ligands 2, 3 or 4 to these MOs is smaller. This leads us to suggest that the better dye performance of [Cu(1)(5)]+ may be associated with improved hole transfer over the halogen of the aryl substituent to the reduced electrolyte.
Conclusions
The synthesis and characterization of [Cu(2)2][PF6] and [Cu(3)2][PF6] have been reported and the spectroscopic and electrochemical properties compared with those of the bromo-analogue [Cu(4)2][PF6]. The preparation of [Cu(5)2][PF6] from [Cu(MeCN)4][PF6] and 5 leads to a mixture of [Cu(5)2][PF6] and [Cu(5)(MeCN)2][PF6] which, in MeCN solution, are in equilibrium at room temperature. The performance of DSCs containing surface-anchored heteroleptic dyes [Cu(1)(2)]+, [Cu(1)(3)]+, [Cu(1)(4)]+ and [Cu(1)(5)]+ depend upon the halo-substituent. Initially, the global efficiencies, η, of the dyes follow the order I > F ≈ Br > Cl. Ripening of the DSCs occurs and after 7 days, the dependence of η on the halo-substituent is I > Cl ≈ F ≈ Br; the highest η is 3.16% for [Cu(1)(5)]+ compared to 7.63% for N719 (a relative efficiency of 41.4% versus N719 set at 100%). Compared to the other halo-substituted dyes, [Cu(1)(5)]+ exhibits an extended spectral response to longer wavelength, with enhanced electron injection. The performance of [Cu(1)(5)]+ is surprising considering the structural simplicity of ancillary ligand 5 and the lack of a peripheral electron donating group. DFT calculations have been used to establish the HOMO and LUMO characteristics of the ground-states of [Cu(1)(2)]+, [Cu(1)(3)]+, [Cu(1)(4)]+ and [Cu(1)(5)]+. The results show that ancillary ligand character is present within the HOMO manifold of [Cu(1)(5)]+, with a dominant contribution from the iodophenyl substituent; corresponding contributions from ligands 2, 3 or 4 to these MOs in [Cu(1)(2)]+, [Cu(1)(3)]+, [Cu(1)(4)]+ is smaller. This suggests that the improved dye performance of [Cu(1)(5)]+ may result from better electron transfer over the halogen of the aryl substituent from the reduced electrolyte.The observation that DSCs containing [Cu(1)(5)]+ give global efficiencies >3% is unexpected and has significant potential in terms of the use of a synthetically very accessible ancillary ligand. The same level of efficiency can be achieved with [Cu(1)(5)]+ assembled using a 1:1 mixture of [Cu(MeCN)4]+ and 5 in place of [Cu(5)2]+. This not only avoids the need to prepare the homoleptic complex, but also prevents the wastage of one equivalent of ancillary ligand. Our next challenge is the optimization of the DSCs with [Cu(1)(5)]+ and related dyes, starting with an investigation of the role of co-adsorbents such as cheno.
Acknowledgements
The European Research Council (Advanced Grant 267816 LiLo), Swiss National Science Foundation and University of Basel are acknowledged for financial support. Roché Walliser and Nik Hostettler are thanked for recording 500 MHz NMR spectra. Dr Biljana Bozic-Weber is acknowledged for helpful discussions and technical support.Notes and references
- N. Alonso-Vante, J.-F. Nierengarten and J.-P. Sauvage, J. Chem. Soc., Dalton Trans., 1994, 1650 Search PubMed.
- G. C. Vougioukalakis, A. I. Philippopoulos, T. Stergiopoulos and P. Falaras, Coord. Chem. Rev., 2011, 255, 2602 CrossRef CAS PubMed and references therein.
- N. Robertson, ChemSusChem, 2008, 1, 977 CrossRef CAS PubMed.
- B. Bozic-Weber, E. C. Constable and C. E. Housecroft, Coord. Chem. Rev., 2013, 257, 3089 CrossRef CAS PubMed.
- B. Bozic-Weber, S. Brauchli, E. C. Constable, S. O. Fürer, C. E. Housecroft and I. A. Wright, Phys. Chem. Chem. Phys., 2013, 15, 4500 RSC.
- B. Bozic-Weber, S. Y. Brauchli, E. C. Constable, S. O. Fürer, C. E. Housecroft, F. J. Malzner, I. A. Wright and J. A. Zampese, Dalton Trans., 2013, 42, 12293 RSC.
- B. Bozic-Weber, E. C. Constable, S. O. Fürer, C. E. Housecroft, L. J. Troxler and J. A. Zampese, Chem. Commun., 2013, 49, 7222 RSC.
- C. J. Martin, B. Bozic-Weber, E. C. Constable, T. Glatzel, C. E. Housecroft and I. A. Wright, J. Phys. Chem. C, 2014, 118, 16912 CAS.
- T. E. Hewat, L. J. Yellowlees and N. Robertson, Dalton Trans., 2014, 43, 4127 RSC.
- K. A. Wills, H. J. Mandujano-Ramirez, G. Merino, D. Mattia, T. Hewat, N. Robertson, G. Oskam, M. D. Jones, S. E. Lewis and P. J. Cameron, RSC Adv., 2013, 3, 23361 RSC.
- A. Colombo, C. Dragonetti, D. Roberto, A. Valore, P. Biagini and F. Melchiorre, Inorg. Chim. Acta, 2013, 407, 204 CrossRef CAS PubMed.
- M. Sandroni, M. Kayanuma, A. Planchat, N. Szuwarski, E. Blart, Y. Pellegrin, C. Daniel, M. Boujtita and F. Odobel, Dalton Trans., 2013, 42, 10818 RSC.
- J. Baldenebro-Lopez, N. Flores-Holguin, J. Castorena-Gonzalez and D. Glossman-Mitnik, J. Photochem. Photobiol., A, 2013, 267, 1 CrossRef CAS PubMed.
- L. N. Ashbrook and C. M. Elliott, J. Phys. Chem. C, 2013, 117, 3853 CAS , erratum: 2013, 117, 11883.
- N. Armaroli, Chem. Soc. Rev., 2001, 30, 113 RSC.
- N. Armaroli, Top. Curr. Chem., 2007, 280, 69 CrossRef CAS.
- M. Sandroni, L. Favereau, A. Planchat, H. Akdas-Kilig, N. Szuwarski, Y. Pellegrin, E. Blart, H. Le Bozec, M. Boujtita and F. Odobel, J. Mater. Chem. A, 2014, 2, 9944 CAS.
- B. Bozic-Weber, E. C. Constable, C. E. Housecroft, P. Kopecky, M. Neuburger and J. A. Zampese, Dalton Trans., 2011, 40, 12584 RSC.
- G. J. Kubas, Inorg. Synth., 1990, 28, 68 CrossRef CAS.
- E. C. Constable, C. E. Housecroft, M. Neuburger, I. Poleschak and M. Zehnder, Polyhedron, 2003, 22, 93 CrossRef CAS.
- Y. Takada, K. Nomura and S. Matsubara, Org. Lett., 2010, 12, 5204 CrossRef CAS PubMed.
- Bruker Analytical X-ray Systems, Inc., APEX2, version 2 User Manual, M86-E01078, Madison, WI, 2006 Search PubMed.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- S. Ito, P. Chen, P. Comte, M. K. Nazeeruddin, P. Liska, P. Péchy and M. Grätzel, Prog. Photovoltaics, 2007, 15, 603 CAS.
- S. Ito, T. N. Murakami, P. Comte, P. Liska, C. Grätzel, M. K. Nazeeruddin and M. Grätzel, Thin Solid Films, 2008, 516, 4613 CrossRef CAS PubMed.
- F. Krönhke, Synthesis, 1976, 1 CrossRef.
- M. Hesse, H. Meier and B. Zeeh, Spektroskopische Methoden in der organischen Chemie, Thieme, Stuttgart, 2005 Search PubMed.
- Y. Jahng, J. Hazelrigg, D. Kimball, E. Riesgo, R. Wu and R. P. Thummel, Inorg. Chem., 1997, 36, 5390 CrossRef CAS.
- D. V. Scaltrito, D. W. Thompson, J. A. O'Callaghan and G. J. Meyer, Coord. Chem. Rev., 2000, 208, 243 CrossRef CAS.
- A. Hernandez Redondo, E. C. Constable and C. E. Housecroft, Chimia, 2009, 63, 205 CrossRef.
- H. J. Snaith, Energy Environ. Sci., 2012, 5, 6513 Search PubMed.
- F. J. Malzner, S. Y. Brauchli, E. Schönhofer, E. C. Constable and C. E. Housecroft, Polyhedron, 2014 DOI:10.1016/j.poly.2014.05.019.
- B. Wenger, M. Grätzel and J.-E. Moser, Chimia, 2005, 59, 123 CrossRef CAS.
- B. Wenger, M. Grätzel and J.-E. Moser, J. Am. Chem. Soc., 2005, 127, 12150 CrossRef CAS PubMed.
- V. K. Thorsmølle, B. Wenger, J. Teuscher, C. Bauer and J.-E. Moser, Chimia, 2007, 61, 631 CrossRef.
- See for example: S. Y. Brauchli, B. Bozic-Weber, E. C. Constable, N. Hostettler, C. E. Housecroft and J. Zampese, RSC Adv., 2014, 4, 34801 RSC.
- B. Bozic-Weber, V. Chaurin, E. C. Constable, C. E. Housecroft, M. Meuwly, M. Neuburger, J. A. Rudd, E. Schönhofer and L. Siegfried, Dalton Trans., 2012, 41, 14157 RSC.
Footnote |
| † CCDC [1000780]. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra06823h |
| This journal is © The Royal Society of Chemistry 2014 |