
Adsorption-photocatalytic degradation of methyl orange over a facile one-step hydrothermally synthesized TiO2/ZnO–NH2–RGO nanocomposite†
Abstract
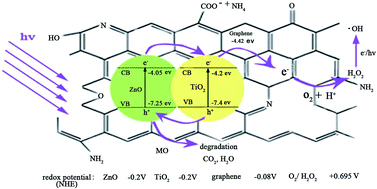
A novel TiO2/ZnO–NH2–reduced graphene oxide (TZ-a-RGO) nanocomposite was successfully prepared using a facile one-step hydrothermal method. The TZ-a-RGO was characterized by X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), Transmission electron microscopy (TEM), Brunauer–Emmett–Teller (BET) surface area analysis and UV-vis absorption spectrophotometry to investigate its structural features. The TZ-a-RGO was used as a catalyst to remove methyl orange (MO) from wastewater, and the results indicated that this catalytic system has a good performance in terms of removal of MO. The adsorption experiments of the TZ-a-RGO followed the pseudo-second-order kinetic model, and the adsorption isotherms were accurately represented by the Langmuir model. The degradation of methyl orange (MO) by TZ-a-RGO fitted well with the Langmuir–Hinshelwood model, and MO removal was obtained through a synergistic effect of adsorption and photocatalysis. The photocatalytic rate of MO over the composites was as high as 8.2 and 3.2 times that over commercial P25 (Degussa) and TiO2/ZnO, respectively. The potential photocatalytic mechanism for the TZ-a-RGO nanocomposite under UV was discussed.
 Please wait while we load your content...
Please wait while we load your content...

