
Chemo-enzymatic synthesis of bicyclic 3′-azido- and 3′-amino-nucleosides†
Abstract
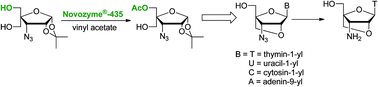
Conformationally locked 3′-azido-3′-deoxythymidine analogues of T, U, A and C containing a 2′-O,4′-C-methylene linked bicyclic furanose moiety has been efficiently synthesized following a greener chemo-enzymatic convergent route. Thus, one of the two diastereotopic hydroxyl groups of 3-azido-3-deoxy-4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-ribofuranose has been regioselectively acetylated using Novozyme®-435 in quantitative yield. The selective enzymatic acetylation can be carried out with the same efficiency using Novozyme®-435 for 10 cycles of reaction. The monoacetylated sugar derivative was converted to bicyclic 3′-azidonucleosides in four steps in overall yields of 60 to 68%. It has been demonstrated that 3′-azido-3′-deoxy-2′-O,4′-C-methylenethymidine can easily be converted into 3′-amino-3′-deoxy-2′-O,4′-C-methylenethymidine in 95% yield, which is an important monomer for the synthesis of therapeutically useful sugar-modified oligonucleotides.
 Please wait while we load your content...
Please wait while we load your content...

