
Fabrication of robust M/Ag3PO4 (M = Pt, Pd, Au) Schottky-type heterostructures for improved visible-light photocatalysis†
Abstract
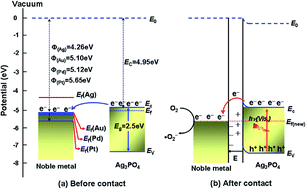
M/Ag3PO4 (M = Pt, Pd, Au) Schottky-type heterostructures were successfully fabricated by a chemical deposition route using NaBH4 as a reduction agent. The structure and optical properties of the as-synthesized samples were characterized by X-ray diffraction, scanning electron microscopy, transmission electron microscopy, X-ray photoelectron spectroscopy, UV-vis diffuse reflectance spectroscopy, and electrochemical techniques. The photocatalytic activity was evaluated by the decomposition of dyes (methyl orange, methylene blue, and rhodamine B) under visible light irradiation (λ > 420 nm). These noble metals as nanoparticles were highly dispersed on the surface of Ag3PO4 polyhedrons. The light absorption of Ag3PO4 in both the UV and visible regions was extensively increased upon noble metal deposition. The photocurrent response over M/Ag3PO4 electrodes was much higher than that of pure Ag3PO4 and followed the decreased order of Pt > Pd > Au. The metallic nanoparticles deposited on Ag3PO4 could promote the transfer of photo-generated electrons, which not only inhibited the recombination of electrons and holes effectively, but also suppressed the photocorrosion of Ag3PO4, leading to a significant increase in photocatalytic activity and stability. On the basis of radical-trapping experiments and the PL technique, h+ and ˙O2− were well-established to correspond to the quick photo-degradation of dyes and a possible mechanism was proposed.
 Please wait while we load your content...
Please wait while we load your content...

