
Development of a highly sensitive and selective spectrophotometric method for the determination of carvedilol in pharmaceutical and urine samples
Masoud Shariati-Rad*,
Mohsen Irandoust and
Sara Sheikhi
Department of Analytical Chemistry, Faculty of Chemistry, Razi University, Kermanshah, Iran. E-mail: mshariati_rad@yahoo.com; Fax: +98 831 4274559
First published on 12th August 2014
Abstract
A simple, sensitive, selective, accurate and cost-effective spectrophotometric method for the determination of CAR in pharmaceutical and urine samples was developed. The method is based on the reaction between carvedilol and nitrite. Factors affecting the reaction were concentration of the reagents, including hydrochloric acid, sodium nitrite and sodium hydroxide. The factors were optimized using a central composite design. In optimum conditions, two calibration curves at 250 and 278 nm were constructed with linear ranges of 0.05–0.20 and 1.5–3.5 mg L−1 and detection limits of 1.8 × 10−4 and 8.1 × 10−4 mg L−1, respectively. The application of the proposed method showed precise and accurate results. In complex urine samples, the method was free from interferences. Comparison with the reported methods for carvedilol determination using statistical characteristics revealed that the proposed method is over all superior.
Introduction
Carvedilol (CAR), 1-(4-carbazolyloxy)-3-[2-(2-methoxy)ethylamino]-2-propanol (Fig. 1), is a nonselective β-adrenergic receptor antagonist and a α1-adrenoceptor blocker. The β1-blockade produces a decrease in heart rate and in the force of contraction of the cardiac muscle.1–3 The FDA first approved CAR in 1995.4 It is an official drug in British and European pharmacopoeias. Recently, it was recognized as an effective agent for the treatment of congestive heart failure.5,6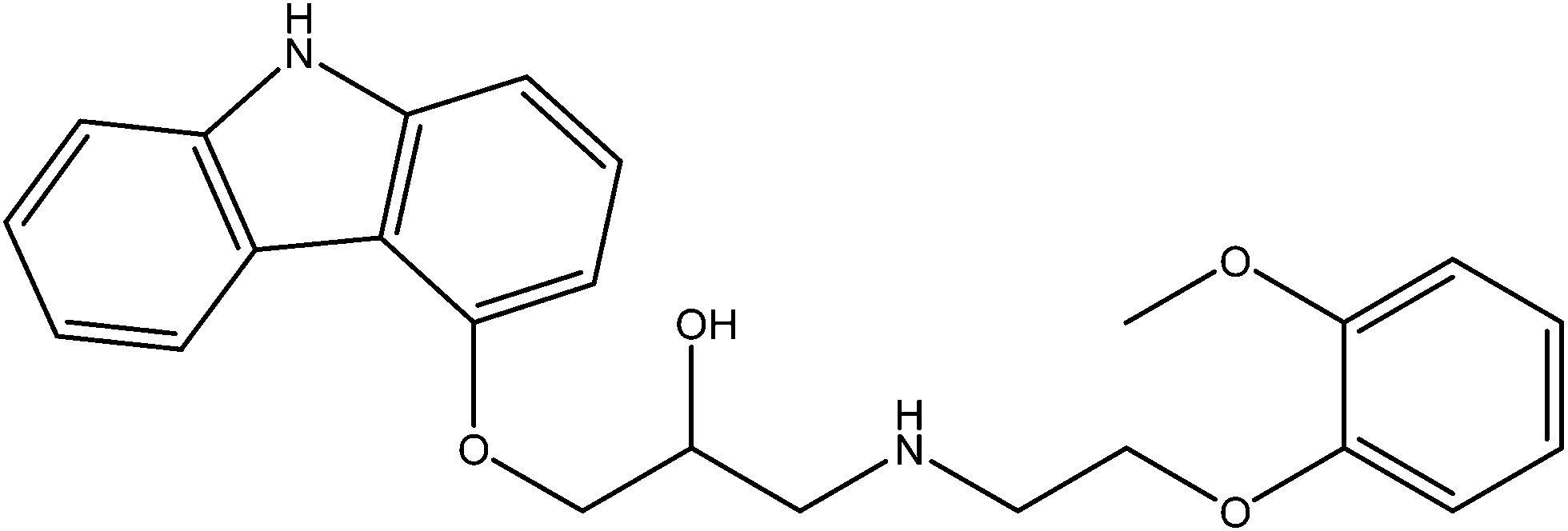 | ||
| Fig. 1 Chemical structure of carvedilol. | ||
Analytical methods such as spectrophotometry,7,8 HPLC,9–12 capillary electrophoresis,13 flow injection spectrofluorimetry,14 differential pulse voltammetry,15 GC-MS,16 liquid chromatography,17 chemiluminometry18 and spectrofluorimetry19,20 have been used for the determination of CAR.
It has been observed that in some of the applications of HPLC11,12 reproducibility and recovery are not satisfactory. Excessive extraction steps and use of organic solvents can also be mentioned as their limitations. Capillary electrophoresis and tandem mass spectrometry for determination of CAR have been reported, but procedures were inconvenient.21,22 A differential pulse voltammetry procedure using a glassy carbon electrode was developed for the analysis of CAR in tablets.13 However, this method has various limitations, including time-consuming sample clean-up, laborious extraction steps, low sensitivity and long run times. Therefore, it is less suitable for routine analysis.
Spectrophotometric methods have been reported for determination of CAR in UV-Vis region.7,8 The charge transfer complex formation systems for analytical determinations7 needs extensive use of toxic organic solvents. In UV-Vis region, interferents are unavoidable, in particular for complex samples like urine. Moreover, these methods cannot be used to analyze complex samples such as urine. An extractive spectrophotometric method has also been used to determine CAR in pharmaceuticals.23 In this procedure, extraction by chloroform, a toxic organic solvent has been proposed.
In regards of the importance of determination of CAR, we developed a sensitive and selective UV-Vis spectrophotometric method for determination of CAR in pharmaceutical and biological samples. For optimization of the reaction conditions, central composite design (CCD) was employed. It was originally developed by Box and Wilson24 and improved by Box and Hunter.25 The proposed method when compared with the reported spectrophotometric methods takes the advantage over the UV spectrophotometric method in terms of selectivity and sensitivity.
Experimental
Apparatus and software
Recording of the absorption spectra in the spectral range of 200–600 nm was performed by an Agilent 8453 UV-Vis spectrophotometer with diode array detector, equipped with 1 cm path length quartz cells. Design and analysis of the central composite experiments were carried out by the MINITAB (Minitab Inc. Release 16.0) statistical package.Reagents and solutions
All the chemicals, i.e., sodium nitrite, sodium hydroxide and hydrochloric acid (Merck KGaA, Darmstadt, Germany) were of analytical grade (>99%) and used as received without any further purification.A 1000.0 mg L−1 standard stock solution of CAR was prepared in absolute ethanol. About 10.00 mg of CAR was accurately weighed and transferred to a 100 mL volumetric flask, ethanol was added to dissolve the drug, and volume was then completed to the mark with ethanol. The stock solution was diluted appropriately to get the working concentrations. Moreover, solutions of sodium nitrite (3.0% (w/v) in water), sodium hydroxide (15.0% (w/v) solution in water) and hydrochloric acid (6.0 M) were prepared for experiments.
Procedure
Two calibration curves were constructed in different concentration ranges of CAR. Accurately measured volumes of standard stock solution of CAR (1.25–7.50 μL and 37.5–87.5 μL) were transferred to separate series of 25 mL volumetric flasks. To each of these 25.0 mL volumetric flasks, volumes of the stock solutions of hydrochloric acid, sodium nitrite and sodium hydroxide were then added such that the final concentrations of these reagents reached 0.432 M, 0.234% and 11.04%, respectively. The volume of the flasks was then filled to the mark with doubly distilled water, the contents were efficiently shaken and the absorbance of the solutions was measured at the wavelengths 250 and 278 nm, respectively. Calibration curves were constructed for CAR by plotting a graph of absorbance versus concentration at wavelengths 250 and 278 nm.Pharmaceutical preparation
From each type of the tablet (Carvedilol 12.5 and Carvedilol 6.25 from Farabi Pharmaceutical Company) ten tablets were pulverized. An accurately weighed quantity of the mixed powder equivalent to one tenth of the weight of a tablet was transferred into a 10 mL volumetric flask and made up to the mark with ethanol. The content was shaken for 30 min, filtered and quantitatively transferred into 10 mL volumetric flasks. The solution was completed to the mark with ethanol. A 0.4 mL aliquot of this solution was transferred into 25 mL volumetric flasks. The reagents were then added according to the manner explained in the Procedure section. The volume was made up to 25 mL with double distilled water, and the absorbance was measured at 250 and 278 nm.Human urine samples
Aliquots of urine (5.0 mL) from informed healthy volunteers were spiked with different concentrations of CAR. In 25 mL volumetric flasks, 1.0 mL of the resulting urine solution was added followed by the addition of the reagents according to the manner explained in the Procedure section. The volume was completed to the mark with double distilled water and the solution was left to stand for 90 min. The absorbance of each solution was measured and the nominal concentration of the drug in urine was determined based on the equation of the calibration curve.Results and discussion
Central composite experimental design and optimization of factors
Experimental design methodology involves simultaneously changing all the factors from one experiment to the next because the factors can influence each other, and the optimum value for one of the factor might be related to the values of the others.In CCD, it is assumed that the central point for each factor is zero and the design is around it symmetrical.26 Factors influencing the studied system and their considered levels for design are shown in Table 1. For a system with three factors (n = 3), CCD consists of 18 experiments. Values of the factors in these 18 experiments and obtained responses are shown in Table 2.
| Factor | Level | ||
|---|---|---|---|
| −1 | 0 | 1 | |
| Parameter values | |||
| X1 (concentration of hydrochloric acid; M) | 0.12 | 0.30 | 0.48 |
| X2 (concentration of nitrite; w/v%) | 0.12 | 0.24 | 0.36 |
| X3 (concentration of sodium hydroxide; w/v%) | 3.0 | 6.0 | 9.0 |
| Experiment no. | Concentration of hydrochloric acid (M) | Concentration of sodium nitrite (w/v%) | Concentration of sodium hydroxide (w/v%) | Response |
|---|---|---|---|---|
| 1 | 0.30 | 0.24 | 11.04 | 0.985 |
| 2 | 0.30 | 0.24 | 0.95 | 0.000 |
| 3 | 0.30 | 0.24 | 6.00 | 0.673 |
| 4 | 0.30 | 0.44 | 6.00 | 0.211 |
| 5 | 0.30 | 0.04 | 6.00 | 0.327 |
| 6 | 0.48 | 0.12 | 9.00 | 0.524 |
| 7 | 0.12 | 0.12 | 9.00 | 0.309 |
| 8 | 0.30 | 0.24 | 6.00 | 0.739 |
| 9 | 0.48 | 0.12 | 3.00 | 0.362 |
| 10 | 0.12 | 0.12 | 3.00 | 0.371 |
| 11 | 0.00 | 0.24 | 6.00 | 0.139 |
| 12 | 0.12 | 0.36 | 3.00 | 0.214 |
| 13 | 0.30 | 0.24 | 6.00 | 0.672 |
| 14 | 0.12 | 0.36 | 9.00 | 0.135 |
| 15 | 0.48 | 0.36 | 9.00 | 0.498 |
| 16 | 0.30 | 0.24 | 6.00 | 0.466 |
| 17 | 0.48 | 0.36 | 3.00 | 0.151 |
| 18 | 0.60 | 0.24 | 6.00 | — |
Analysis of variance (ANOVA) of the performed experiments (Table 2) is given in Table 3. The response of experiment no. 18 in Table 2 was noisy and higher than the scale of the spectrophotometer, and it was not possible to assign an exact response for this experiment. Therefore, the response of this experiment was not considered in ANOVA.
Among the linear terms, the concentration of hydrochloric acid (X1) is significant, and among the squared ones, X2X2 (X2 is concentration of sodium nitrite) is significant (see Table 3). None of the interaction terms is significant at the 95% confidence level.
In order to gain insight about the effect of each factor, response surfaces were constructed. These surfaces which indicate the variations of response with two factors are shown in Fig. 2. For obtaining each surface, the third factor is kept at its central level. From the sign of the coefficients in Table 3, it is clear that in the higher values of factors X1 and X3, the response should be larger. This can also be observed from different panels of Fig. 2. Moreover, from Fig. 2, it can be concluded that in the intermediate amounts of factor X2 (concentration of nitrite) the response is higher. Curvatures in the surfaces, when factors X1 and X2 (concentration of hydrochloric acid and concentration of sodium nitrite, respectively) vary (Fig. 2) indicate some self-interaction (squared term) among these factors. For squared terms of these factors, p values are very low and close to the critical value of 0.05 (see Table 3). It can be observed from Fig. 2b that some interaction exists between factors X1 and X3 (concentration of hydrochloric acid and concentration of sodium hydroxide, respectively). From Table 3 it can be found that the p value of the interaction X1X3 is lower than the corresponding value for X1X2 and X2X3 interactions.
 | ||
| Fig. 2 Variation of response with (a) concentration of nitrite (X2) and concentration of hydrochloric acid (X1), (b) concentration of sodium hydroxide (X3) and concentration of hydrochloric acid (X1) and (c) concentration of nitrite (X2) and concentration of sodium hydroxide (X3). Maximum of each surface and its value has been shown. | ||
The response surface plots in Fig. 2 show that all of the response surfaces have maximum points. Therefore, response surface optimization could be used. Results of the response optimization showed that the response should be at maximum with 0.432 M hydrochloric acid, 0.234% (w/v) sodium nitrite and 11.04% (w/v) sodium hydroxide. Therefore, a high concentration of hydrochloric acid and concentrations near the center of the design for sodium nitrite are suitable for the studied reaction.
Absorption spectra
The spectrum of CAR in double distilled water (pH = 7.0) and spectrum of its reaction product with nitrite under optimum conditions are shown in Fig. 3. | ||
| Fig. 3 Absorption spectra of CAR with concentration of 2.0 mg L−1 (a) in water pH = 7.0 and (b) after reaction with nitrite under optimum conditions. | ||
As seen from Fig. 3, CAR shows weak absorption in the range of 190–350 nm with a maximum absorption at 240 nm. These low absorbances result in very low sensitivity for CAR determination. However, after reaction with nitrite27,28 under optimum conditions, a product is formed that shows an intense spectrum that extends to about 500 nm. The maximum of the spectrum is located at 250–280 nm (Fig. 3b) and the corresponding shoulder is located at about 370 nm. This indicates the high sensitivity of the proposed method for the determination of the studied drug.
Based on the reaction between indole derivatives and nitrite under the same conditions reported in the literature,27,28 the possible reaction between CAR and nitrite can be shown as in Scheme 1.
 | ||
| Scheme 1 The possible reaction between CAR and nitrite under optimum conditions. | ||
In the first step, CAR with nitrous acid (HNO2) undergoes diazotization. In the next step, the unstable diazonium group is replaced by a hydroxyl group in aqueous medium. The formed hydroxyl compound now has a free para-position that can react with nitrous acid HNO2 and give a compound, which after dissociation of the hydroxyl group in alkaline medium is responsible for the formation of yellow color.
Analytical data
Under the optimized experimental conditions, adherence to Beer's law was studied by measuring the absorbance values of the solutions with varying prepared drug concentrations as discussed in the Procedure. Two calibration curves were constructed. The calibration curves were linear at concentration ranges of 0.05–0.20 and 1.5–3.5 mg L−1 at wavelengths 250 and 278 nm, respectively. Calibration curves are shown in Fig. 4. Statistical parameters of the calibration curves were calculated and reported in Table 4. The optical characteristics such as molar absorptivities of the products are also mentioned in Table 4. The high values of molar absorptivity and slope of the calibration curves, and the low values of DL indicate the high sensitivity of the proposed method. However, statistical data in Table 4 imply that the calibration curve at 250 nm is about four times more sensitive than the one at 278 nm. The standard errors of the parameters of calibrations are also significantly low. The linearity of the calibration curve is validated by the high value of the correlation coefficient (which were close to unity) of the calibration curve.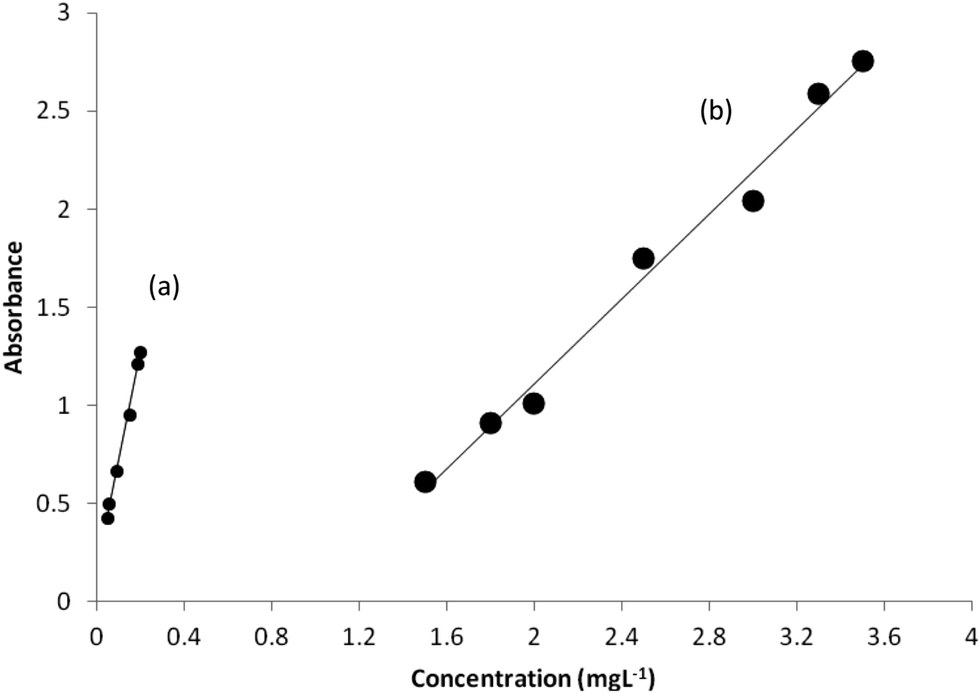 | ||
| Fig. 4 Calibration curves constructed with the absorbances at (a) 250 nm and (b) 278 nm under optimum conditions. | ||
| Parameters | Calibration curve 1 | Calibration curve 2 |
|---|---|---|
| a Calculated as DL = yB + 3sB, where yB is the signal of the blank (intercept of the calibration curve) and sB is the standard deviation of the blank.29 | ||
| λmax (nm) | 250 | 278 |
| Molar absorptivity of the product (L mol−1 cm−1) | 3.41 × 106 | 3.19 × 105 |
| Linear range (mg L−1) | 0.05–0.20 | 1.5–3.5 |
| Intercept of calibration curve | 0.159 | 1.051 |
| Slope of calibration curve | 5.423 | 1.081 |
| Standard error of intercept | 0.028 | 0.141 |
| t statistics of intercept | 21.18 | 7.45 |
| Standard error of slope | 0.237 | 0.054 |
| Standard error of regression | 0.027 | 0.103 |
| t statistics of slope | 22.88 | 20.02 |
| Correlation coefficient | 0.998 | 0.994 |
| Detection limit (DL)a | 1.76 × 10−4 | 8.12 × 10−4 mg L−1 |
Application of the proposed method for pharmaceutical preparation
The constructed calibration curves were examined for the determination of CAR in tablets and biological samples.Under the optimized experimental conditions, the proposed method was used to determine CAR in commercial tablets. In this case, because the solution resulted by dissolving commercial tablets have higher concentrations of CAR, the second calibration curve was used for the prediction of concentration. The results of determination of CAR in two commercial tablets have been mentioned in Table 5. The percentage of relative standard deviation values (RSD%) are acceptable (below 6%). Accuracy was evaluated as percentage relative error in prediction (RE%). As suggested by the RE% values in Table 5, the proposed method is highly accurate with values about 1% and lower.
Application of the proposed method for urine samples
The high selectivity and sensitivity of the proposed method can allow the determination of CAR in human urine samples. The results of the analysis of the urine samples have been summarized in Table 6. The data obtained by the method indicated that the percent relative error (RE%) and percentage relative standard deviation (RSD%) values are satisfactory. The results, in particular, are excellent for higher concentrations in which the first calibration curve is employed. Very low percent relative errors indicate that the method is selective. It is known that urine is a complex sample having matrix effects and unknown spectral interferences. However, in this application, the results indicate that the method is highly selective.Interference study
In the application of the proposed method to pharmaceutical and in particular biological samples, it is important to test its selectivity towards different potential interferents. Several species that can occur in the real samples along with the drug were investigated. The tolerance limit of the potentially interfering species was taken as its maximum amount causing an error of ≥±5% during the determination of the drug. The anions were used as sodium and potassium salts and the cations as chlorides. The results are shown in Table 7.| Foreign ions | Added as | Tolerance limit (mg L−1) |
|---|---|---|
| K+ | KCl | 13 |
| Ca2+ | CaCl2·2H2O | 20 |
| Mg2+ | MgCl2·4H2O | 4.5 |
| NO3− | NaNO3 | 138 |
| SO42− | Na2SO4 | 152 |
| Paracetamol | — | 45 |
| Cephalexin | — | 10 |
| Diphenhydramine | — | 25 |
Nitrate and sulphate as typical anions do not interfere with the determination of CAR. As typical drugs, paracetamol, cephalexin and diphenhydramine were tested, and it was observed that they do not interfere in the determination of CAR. Potassium and calcium do not significantly interfere. The only interferent was magnesium ion, which can be masked along with other cations by EDTA.
Comparison with the reported methods
In Table 8, the main characteristics of the reported methodologies for the determination of CAR have been mentioned. The detection limit of the proposed method is the lowest, compared with the spectrophotometric methods reported in Table 8. The high values of molar absorptivity and low values of DL indicate the high sensitivity of the proposed method. The lowest DL reported for CAR determination has been obtained by gas chromatography with mass spectrometric detection (GC-MS). However, the proposed method in this work for determination of CAR is about two times more sensitive than GC-MS. The values of RSD% and RE% of the proposed method are low, and the linear ranges of the method presented here are satisfactory and relatively wide. In this method, we broadened the linear ranges of the calibration. Moreover, the spectrophotometric methods developed have potentials limited to simple samples, such as pharmaceuticals.| Instrumental methodology | Experimental details | DL (mg L−1) | Samples | Ref. |
|---|---|---|---|---|
| a p-Dimethylaminobenzaldehyde.b Ninhydrin.c Acetaldehyde.d Binding constant.e N-Methyl-N-trimethylsilyltrifluoroacetamide.f N-Methyl bis-trifluoroacetamide.g o-Desmethyl carvedilol.h 4′-Hydroxyphenyl carvedilol. | ||||
| Spectrophotometry | Method A: reaction of CAR with PDAB,a pH = 4, λmax = 601 nm, molar absorptivity = 0.92 × 103 L mol−1 cm−1. Method B: the charge transfer complex formation of CAR with p-chloranil; λmax = 662 nm, molar absorptivity = 0.257 × 104 L mol−1 cm−1 | 1.65, 0.6626 | Pharmaceuticals | 7 |
| Spectrophotometry | (1) Reaction of CAR with NINb in basic medium, λmax = 402 nm, molar absorptivity = 2.57 × 104 L mol−1 cm−1. (2) Reaction of CAR with AAc in the presence of sodium nitroprusside in basic medium, λmax = 558 nm, molar absorptivity = 1.617 × 104 L mol−1 cm−1 | 0.139, 0.157 | Pharmaceuticals | 8 |
| HPLC | Brownlee C8 column, isocratic elution and on-line deproteination | 8 × 10−4 (LOQ) | Human plasma | 10 |
| Flow injection spectrofluorimetry | λex = 286 nm, λem = 341 nm, sampling rate: 30 samples per h, KBd = 3.2 × 102 L mol−1 | 1.47 × 10−3 | Pharmaceuticals | 14 |
| Gas chromatography-mass spectrometry | Target compounds were extracted using liquid–liquid extraction. The extracts were completely derivatized with MSTFAe and MBTFAf and analyzed by GC-MS using an Ultra-2 ((5%-phenyl)-methylsiloxane) column | o-DMCg 3 × 10−4, 4-HPCh 7.5 × 10−4 | Human urine | 16 |
| Liquid chromatography | Protein precipitation with methanol, mobile phase consisted of acetonitrile-30 mM potassium dihydrogenphosphate buffer, pH = 2 (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 v/v). Column: develosil 3 μm ODS 100 × 4.6 mm I.D. :70 v/v). Column: develosil 3 μm ODS 100 × 4.6 mm I.D. |
1.3 × 10−3 (LOQ) | Human plasma | 17 |
| Spectrofluorimetry | Reaction of CAR with 1-dimethylaminonaphthalene-5-sulphonyl chloride in the presence of mixture (acetone–0.5 M sodium carbonate, 3:2), pH = 10, λem = 445 nm, λex = 350 nm |
1.9 × 10−3 | Human plasma | 19 |
| Chemiluminometry | Reaction: oxidation of luminol by hypochlorite. Multipumping flow system multiple solenoid actuated μ-pumps | 3.53 × 10−3 | Pharmaceuticals | 18 |
| Spectrophotometry | Reaction of CAR with nitrite. Λmax = 250 nm, molar absorptivity = 3.41 × 106 L mol−1 cm−1. Λmax = 278 nm, molar absorptivity = 3.19 × 105 L mol−1 cm−1 | 1.76 × 10−4, 8.12 × 10−4 | Human urine | This work |
Conclusions
A simple, sensitive, rapid and cost-effective spectrophotometric method was developed and validated for the determination of CAR. The reagent utilized in the proposed method is cheap and readily available, and the procedure does not involve any critical reaction conditions or tedious sample preparation. The method is more selective and sensitive than many of the reported spectrophotometric methods. Moreover, its analytical characteristics are superior over expensive methods like GC-MS and HPLC for the determination of CAR. This method can be used as a general method for the determination of CAR in bulk powder, dosage forms and biological samples. The method has many advantages over the separation techniques, such as HPLC, which includes reduced cost and speed with high accuracy. Hence, the method can be used in routine analysis of drugs in quality control laboratories.References
- R. R. Ruffolo, M. Gellai, J. P. Hieble, R. N. Willette and A. J. Nichols, Eur. J. Clin. Pharmacol., 1990, 38, S82 CrossRef CAS.
- A. J. Nichols, M. Gellai and R. R. Ruffolo, Fundam. Clin. Pharmacol., 1991, 5, 25 CrossRef CAS PubMed.
- C. de Mey, K. Breithaupt, J. Schloos, G. Neugebauer, D. Palm and G. G. Belz, Clin. Pharmacol. Ther., 1994, 55, 329 CrossRef CAS.
- B. T. Vanderhoff, H. M. Ruppel and P. B. Amsterdam, Am. Fam. Physician, 1998, 58, 1627 CAS.
- T. L. Yue, H. Y. cheng, P. G. Lysko, P. L. Mckenna, P. Feuerstein, J. L. Gu, K. A. Lysko, L. L. Davis and G. Feuerstein, J. Pharmacol. Exp. Ther., 1992, 263, 92 CAS.
- K. Nakamura, K. Kusano, Y. Nakamura, M. Kakishita, K. Ohta, S. Nagase, M. Yamamoto, K. Miyaji, H. Saito, H. Morita, T. Emori, H. Matsubara, S. Toyokuni and T. Ohe, Circulation, 2002, 105, 2867 CrossRef CAS.
- D. N. Shetty and B. Narayana, ISRN Spectrosc., 2012, 2012, 1 Search PubMed.
- T. V. Sreevidy and B. Narayana, Indian J. Chem. Technol., 2009, 16, 74 Search PubMed.
- Z. Talebpour, M. Taraji and N. Adib, J. Chromatogr. A, 2012, 1236, 1 CrossRef CAS PubMed.
- G. Lamprecht and K. Stoschitzky, Chromatographia, 2004, 59, 551 CAS.
- N. Hokama, N. Hobara, H. Kameya, S. Ohshiro and M. Sakanashi, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 732, 233 CrossRef CAS.
- F. Behn, S. Laer and H. Scholz, J. Chromatogr. Sci., 2001, 39, 121 CAS.
- L. Clohs and K. M. McErlane, J. Pharm. Biomed. Anal., 2001, 24, 545 CrossRef CAS.
- R. A. Silva, C. C. Wang, L. P. Fernandez and A. N. Masi, Talanta, 2008, 76, 166 CrossRef CAS PubMed.
- A. Radi and T. Elmogy, Il Farmaco, 2005, 60, 43 CrossRef CAS PubMed.
- S.-W. Myung and C.-H. Jo, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 822, 70 CrossRef CAS PubMed.
- P. Ptacek, J. Macek and J. Klima, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 789, 405 CrossRef CAS.
- C. K. Pires, K. L. Marques, J. L. M. Santos, R. A. S. Lapa, J. L. F. C. Lima and E. A. G. Zagatto, Talanta, 2005, 68, 239 CrossRef CAS PubMed.
- L. El sayed, A. Fattah, T. A. Mohamed and E. A. Taha, Chem. Ind. Chem. Eng. Q., 2010, 16, 31 CrossRef.
- L. X. Xu, N. Hui, L. Y. Ma and H. Y. Wang, Spectrochim. Acta, Part A, 2005, 61, 855 CrossRef PubMed.
- G. Egginger, W. Lindner, C. Vandenbosch and D. L. Massart, Biomed. Chromatogr., 1993, 7, 277 CrossRef CAS PubMed.
- L. Clohs and K. M. McErlane, J. Pharm. Biomed. Anal., 2001, 24, 545 CrossRef CAS.
- J. Verma and H. Syed, Indian J. Pharm. Sci., 2007, 69, 303 CrossRef CAS PubMed.
- G. E. P. Box and K. B. Wilson, J. Roy. Stat. Soc. B, 1951, 13, 1 Search PubMed.
- G. E. P. Box and J. S. Hunter, Ann. Math. Stat., 1957, 28, 195 CrossRef.
- R. G. Brereton, Chemometrics: Data Analysis for the Laboratory and Chemical Plant, John Wiley, 2003, pp. 76–84 Search PubMed.
- K. K. Verma and A. Jain, Talanta, 1988, 35, 35 CrossRef CAS.
- N. Jie, J. Yang and F. Meng, Talanta, 1993, 40, 1009 CrossRef CAS.
- Statistics and chemometrics for analytical chemistry, ed. J. N. Miller and J. C. Miller, Pearson Education Limited, London, 5th edn, 2005, p. 114 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |