
Dual drug loaded vitamin D3 nanoparticle to target drug resistance in cancer†
Abstract
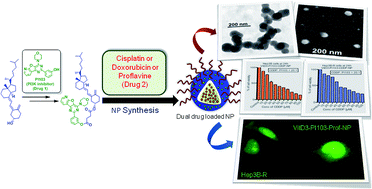
Overcoming drug resistance is one of the most challenging problems in cancer chemotherapy. Drug cocktails can overcome the drug resistance. However, multiple drug combinations lead to multifold increment of off-target toxicity, as well as the delivery of the required therapeutic amount of combined drugs remains problematic. To address these problems, we have developed a sub 200 nm vitamin D3 nanoparticle, which can contain a rational combination of dual drugs (PI103 and cisplatin or doxorubicin or proflavine). The size, shape and morphology of these dual drug containing vitamin D3 nanoparticles were characterized by DLS, FESEM, AFM and TEM. The nanoparticles released the dual drugs in high quantity at pH = 5.5 compared to pH = 7.4 in a slow and sustained manner over 72 h with stability over 15 days at 37 °C, as well as 4 °C. These dual drug loaded nanoparticles induced increased cell death in human hepatocellular carcinoma, Hep3B cells at 24 h compared to monotherapy; moreover, they were effective against cisplatin-resistant cells (Hep3B-R) as well. VitD3–PI103–CDDP-NP and vitD3–PI103–Dox-NP showed cytotoxicity by inducing apoptosis through DNA damage. Furthermore, vitD3–PI103–CDDP-NP showed considerably improved efficacy in 5-fluorouracil (5-FU) resistant Hep3B–5FU-R cells; its activity was even better compared to 5-FU. Finally, vitD3–PI103–proflavine-NP internalized into Hep3B-R cells considerably faster (within 3 minutes) compared to Hep3B cells, as visualized by fluorescent microscopy. Therefore, these dual drug loaded nanoparticles can successfully overcome the trauma of drug resistance and have the potential to be applied into the clinics for improved cancer therapeutics.
 Please wait while we load your content...
Please wait while we load your content...

