
Horseradish peroxidase (HRP) as a tool in green chemistry
Guido R. Lopes,
Diana C. G. A. Pinto* and
Artur M. S. Silva*
Department of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal. E-mail: artur.silva@ua.pt; diana@ua.pt
First published on 6th August 2014
Abstract
Horseradish peroxidase (HRP) is a well-known and commercially available enzyme that has been used over the last years in a wide range of applications from which one can emphasize its leading role in organic synthesis with direct influence in the medical field. This review highlights the versatility of this enzyme, pointing out the work developed in biocatalysis research that provide new interesting products supported in green sustainable methodologies. Therefore, the goal is not to deeply explore the issues related to the mechanistic concepts of the reactions, but to use some selected examples to show the broad range of compounds that can be obtained in reactions with HRP, evidencing its use mainly in organic synthesis.
1. Introduction
The classical chemical approach for the synthesis of a molecule involves the use of an initial specific compound and its transformation into new derivatives. Additionally, it is expected that the new compounds present improved biological activities or industrial applications. In the last decades, biotransformations have been developed as an alternative to this classical approach.1 Indeed some authors compare these different approaches pointing out the disadvantages of the classical organic chemistry. However, in our opinion bio-based chemistry should not be seen as a competitor or a stopgap for non-enzymatic chemistry. In fact, biotransformations offers the inherent diversity of natural world as a great advantage being this approach complementary to the classical one in the demand for novel and biologically or industrial active molecules. Peroxidases (PODs) comprise a group of enzymes that catalyse a variety of oxidative transformations using hydrogen peroxide or other peroxides as oxidants. They have been successfully used in a large number of their potential applications, being horseradish peroxidase (HRP) one of the most studied.2 In this review, we will discuss how the use of HRP can contribute to the development of more efficient and environmentally friendly chemical transformations. It should be pointed out that due to the capability of HRP to catalyse several types of reactions there are thousands of research reports on this topic. Herein, as a logical consequence, this review article cannot give a comprehensive overview of all the cases reported on the literature. We will instead present the diversity of sustainable synthetic reactions and methodologies, describing the compounds used as substrates and/or the high value of products that can be obtained with HRP. The examples described thereafter highlight the versatility and benefit that can be successfully achieved if this special enzyme is employed in bio-based synthetic organic transformations. HRP is able to perform a relatively vast set of reactions, although its efficiency varies in each type of reaction which consequently led to a different amount of publications concerning each field. Even though we will focus on polymerization and coupling reactions as well as hydroxylation and oxygen-transfer reactions, this enzyme is also able to perform N- and O-dealkylations which could be interesting considering that normally harsh conditions are used in this transformations.3,4 Oppositely, we can point out that the poor capability of HRP to catalyse epoxidation reactions led the researchers to develop mutants of the enzyme whose action is more satisfactory.3,52. HRP: structure and stability
Horseradish peroxidase is an oxidoreductase that had been extensively studied along the years and which has shown to be a useful tool in biotechnology. In fact, this enzyme has been used in a huge variety of applications in environmental6 (e.g. wastewater remediation) and biological fields7 (e.g. DNA sensors). Moreover, due to its high thermal resistance HRP is frequently used in food industry to control the thermal processing and food stability.8 Although the root of horseradish contains several peroxidase isoenzymes the knowledge about horseradish peroxidase comes mainly from studies with isoenzyme C, the most abundant one, and due to the production of its corresponding recombinant enzyme.3 Dates back to 1810 the first publication about a reaction catalysed by HRP, the oxidation of 2,5-di(4-hydroxy-3-methoxyphenyl)-3,4-dimethylfuran.9Structurally, HRP isoenzyme C consists of 308 amino acid residues, 4 disulphide bridges between cysteine residues as well as a heme group [iron(III) protoporphyrin IX] and two calcium atoms.3 Moreover, this glycoprotein with 44 kDa has a carbohydrate content of nearly 18–22% and although there is a fraction of β-sheet composition, the structure of HRP is predominantly α-helical.3,10 Details of enzyme structure as well as the somewhat complex catalytic mechanism of HRP action were well described in the literature.3,11 It can be highlight that the different catalytic intermediates can be distinguished from the native structure by UV-vis absorption spectra.12 Peroxidases reactions can be simply summarized in the following equation:
| 2RH + H2O2 → 2R˙ + 2H2O | (1) |
HRP stability, or in a general perspective, enzymes stability is crucial for the application of biocatalysts in industrial domains. In fact, several studies have been made to evaluate HRP stability taking into account parameters as the solvent, the pH or the temperature since like any other chemical catalyst its performance depends on it (Fig. 1).13 Despite being seen as unstable and delicate structures, enzymes can be resistant as any chemical catalyst if used in an appropriate way. It should be noted that the reaction conditions can differ dramatically from the cells conditions, since it may involve high temperatures, extreme pH values, high substrate and/or product, oxidant and organic solvent concentrations (Fig. 1). Enzymes can tolerate these conditions during minutes or hours, but when thinking in industrial applications and in continuous processes it is desirable that reaction conditions can be tolerated during larger periods of time. The use of mixtures water and organic solvents as medium to perform enzymatic reactions has received special attention.14 The partial or total replacement of water as reaction medium by an organic solvent enable that hydrophobic substrates can be converted more efficiently, that higher yields are obtained and that the thermodynamic equilibrium of hydrolytic reactions can be directed through the synthesis pathway.15 In enzymatic catalysis, the water has a primordial role since it allows the flexibility of the active site. However the organic solvent content may not significantly disturb the enzyme–substrate affinity whereby the water content is sufficient to the enzymatic process. However the enhance of organic solvent content in reaction medium can lead to enzyme(s) inactivation due to reversible modifications of protein structure, while prolonged incubations can lead to its irreversible inactivation.14 Santucci et al. referred that the activity of HRP in DMSO is quite similar to the observed in aqueous medium remaining its compact tertiary structure and substantial integrity of the microenvironment of its active site in a 60% (v/v) DMSO solution.13a In higher percentages the organic solvent goes through the protein structure affecting the enzymatic stability and activity. In these conditions the cleavage of hydrogen bonds in the iron core occurs which lead to the unfolding of the protein and to the release of the prosthetic group from the protein matrix. In an interesting study concerning modification of HRP to increase its thermal and organic solvent tolerance Liu et al. verified that native HRP still remains 60% of activity in a solution with 50% (v/v) DMSO during 1 h and at room temperature.16 On the other hand, Azevedo et al. reported that at 50% (v/v) DMSO the activity of HRP is almost nil.14 The authors concluded that denaturation process is only observed when the concentration of DMSO is higher than 20% while a significant decrease occurs with higher percentages. Although the conditions used are not exactly the same (which influences the result) these somewhat contradictory results obtained with the same organic solvent (the best organic solvent for enzymatic reactions) elucidate that this is a primordial problem when dealing with enzymatic reactions. In fact, already in 1968 a study showed that the activity of HRP in various solvents, such as formic acid, methanol or glycerol was similar to its activity in water.17
 | ||
| Fig. 1 Representation of the enzymatic conditions adjustment. | ||
Temperature is also an important operation factor in enzymatic reactions, since it affects the viscosity of reactions medium, the interactions responsible for the structural integrity of the enzyme and the solubility of the substrate and/or the product of the reaction. The velocity of the reaction increases in general with increasing the temperature, but this also leads to the loss of interaction forces which result in loss of activity or even enzyme inactivation. The denaturation of proteins induced by temperature may cause aggregation phenomena at a temperature range of 30–80 °C.18 Theoretically enzymes work better at physiological temperatures; however some of them have a better performance at high temperatures as the ones that come from thermophiles.19 The development of biocatalytic processes usually involves a prior investigation to determine the optimum operating temperature for enzymatic reaction(s). In a study concerning HRP immobilization, Temoçin et al. concluded that the activity of free enzyme has its maximum at pH 8 and a temperature of 45 °C.20 Furthermore, the free enzyme lost about 72% of activity in 60 days at pH 7 and 4 °C as well as its activity was reduced about 40%, 60% and 65% if it was placed in solutions of 10% acetone, acetonitrile or methanol, respectively. The denaturation process seems to be associated with the dissociation of the heme group which may be dependent of the reaction pH and occur faster a pH values lower than 5. In fact, it was reported the complete separation of prosthetic group at pH 2.4 and 25 °C as well as the severe change of secondary structure of the enzyme at 74 °C (melting temperature) which is also associated with heme group release.8,13d
The stability of enzyme's structure greatly depends on the molecular interactions (hydrogen bonds and electrostatic) that enable their mechanisms of action. Thus, enzymes performance is closely related to the reaction media pH due to the influence of this factor in the referred interactions. Furthermore, enzymes have many polar amino acids in the outer surface that can be protonated or deprotonated depending on the pH of the environment. In general, enzymes show their maximum of activity over a pH range of 5–9,19 near the physiological pH, although as in the case of temperature there are some exceptions that prefer extremes values of pH. In laboratory experiments, the pH of a reaction is usually maintained with the use of buffer solutions. These solutions can interfere with the velocity of the reaction due to the interaction of buffer and enzyme molecules and influence the solubility of the substrates. The pH is an important factor to maintain the stability and the maximum activity of the enzyme and the problem arises when optimum pH values for both conditions differ, which hamper the implementation of the process.18 The buffer content in enzymatic reactions also affects the course of the reactions processes by changing enzymes properties. In fact, Haifeng et al. showed that HRP thermal stability was influenced by buffer content.13b The data obtained allow inferring that the presence of sodium phosphate accelerated the denaturation process of the enzyme and reduced its thermal stability. The literature shows that thermal and organic solvent tolerance may be increased by chemical modification of the enzyme (maleic, citraconic and phthalic anhydride) related to changes in HRP conformation.16,21
A large number of reports discuss the role of H2O2 in enzymes activity and stability, namely peroxidases, as well as the consequences of its inappropriate use. In fact, the molecular mechanism concerning the inactivation of peroxidases by H2O2 is quite complex due to the diverse reactions that can occur.22 The exposure of HRP to a high concentration of H2O2 may lead to an irreversible suicide inactivation with the formation of a highly reactive radical that may instigate the destruction of the prosthetic group.22c,f In fact, it has been studied the ratio between the amount of enzyme and H2O2 to be used in enzymatic reactions to avoid the inactivation of HRP.13c,22e,23 However, the proportion determined in some reports as one which causes the total loss of activity is the same that allows the maintenance of activity at considerable levels in others. Since the use of H2O2 in substantial concentration leads to irreversible inactivation of peroxidases, it can be added continuously to the reaction to keep low its concentration. Moreover even the stability of the substrate or of the product(s) formed may be affected by H2O2 which evidence that its addition requires caution. The huge number of reports concerning enzyme catalysis reactions present in literature shows that many authors choose to use enzymes immobilized. Its characteristics and benefits to biocatalysis are the subject of several reviews.24 In fact, immobilization of enzymes can be quite beneficial since under operational conditions some enzymes are unstable and can lose catalytic activity as well as solve the problem of recovery the enzyme at the end of the process, which is important taking into account its industrial application. Besides enzyme stability the immobilization process can even improve the activity as well as the selectivity of enzymes.25 Some recent reports clearly show that immobilization of the enzyme can be a valuable strategy when dealing with HRP catalysed reactions. However, immobilization step may also lead to loss of enzyme activity.26 In fact, the immobilization of enzymes may be related with processes of adsorption, ionic binding or covalent attachment to a carrier as well as cross-linking of enzymes molecules or entrapment into gels, polymers or reversed micelles. The diversity of linkages is well demonstrated by the diversity of examples present in the literature.26b Recent examples that cover HRP reactions show that the immobilization process may be beneficial.27 The problem of suicide inactivation of HRP (and generally peroxidases) has led to the use of site-directed mutagenesis as well as directed evolution techniques in order to modify the enzyme and circumvent this problem.23b,28 Besides this problem these approaches are helpful to improve catalytic activity, change substrate specificity as well as stereoselectivity which seems to make them more and more necessary in biotransformation processes being this already cover elsewhere.29 Other strategy to avoid the suicide inactivation of HRP by H2O2 is the immobilization of multi-enzyme systems that allows in situ production of hydrogen peroxide although some undesirable by-products may be formed.30
3. Enzymatic polymerization
Nowadays, the production of new materials depends greatly on the synthesis of polymers and one of the primordial utilization of enzymes is in the synthesis of polymers as highlighted by several excellent reviews.31 In fact, the enzymatic polymer synthesis offers interesting advantages over conventional methods as mild reaction conditions, without the use of toxic reagents (e.g. formaldehyde) and could enable the synthesis of new polymeric materials that are not possible to obtain with those traditional methods.32 It should be noted that over the years a huge amount of research concerning polymerization reactions using HRP as catalyst has been made and published. However, particularly interest has been given to phenol which is the most important phenolic compound in industrial fields.33 Moreover, the enzymatic polymerization of phenol derivatives with peroxidases is known to be chemoselective when more than two reactive groups are present in the phenol derivative.34 More than twenty five years ago, Dordick and co-workers reported the HRP catalysed polymerization of phenols in non-aqueous media.35 This enzyme polymerizes some phenols in mixtures of water with organic solvents, such as 1,4-dioxane, acetone and DMF. Some polymers were prepared on a gram scale having high melting points and average molecular weights from 400 to 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da depending on the reaction conditions and the phenol derivative used. In fact, during the 1990s various studies were carried out using substituted phenols 1 as monomers and HRP as catalyst (Table 1) in aqueous/organic media or reverse micellar solutions, which demonstrates that this enzyme can efficiently produce polymeric structures.36,37
000 Da depending on the reaction conditions and the phenol derivative used. In fact, during the 1990s various studies were carried out using substituted phenols 1 as monomers and HRP as catalyst (Table 1) in aqueous/organic media or reverse micellar solutions, which demonstrates that this enzyme can efficiently produce polymeric structures.36,37
| R1 | R2 | R3 | Phenol derivative | Reference |
|---|---|---|---|---|
| H | H | H | Phenol | 36 |
| H | CH3 | CH3 | 3,4-Dimethylphenol | 36d |
| CH3 | H | H | 2-Methylphenol | 36d, 37d |
| OH | H | H | 2-Hydroxyphenol | 36d, 37b |
| CH2OH | H | H | 2-(Hydroxymethyl)phenol | 36d |
| OCH3 | H | H | 2-Methoxyphenol | 36d |
| H | CH3 | H | 3-Methylphenol | 36d, 37a,d |
| H | OCH3 | H | 3-Methoxyphenol | 36d, 37a |
| H | C6H5 | H | 3-Phenylphenol | 36d, 37a |
| H | OC6H5OC6H5 | H | 3-(3-Phenoxyphenoxy)phenol | 36d |
| H | C2H5 | H | 3-Ethylphenol | 37a |
| H | Cl | H | 3-Chlorophenol | 37a |
| H | Br | H | 3-Bromophenol | 37a |
| H | H | CH3 | p-Methylphenol | 36d, 37c,d |
| H | H | C2H5 | p-Ethylphenol | 37c, e, f ,g |
| H | H | C3H7 | p-n-Propylphenol | 37c |
| H | H | C3H7 | p-i-Propylphenol | 37c |
| H | H | C4H9 | p-n-Butylphenol | 37c |
| H | H | C4H9 | p-s-Butylphenol | 37c |
| H | H | C4H9 | p-t-Butylphenol | 37c |
| H | H | C5H11 | p-n-Pentylphenol | 37c |
| H | H | C6H13 | p-n-Hexylphenol | 37c |
| H | H | C7H15 | p-n-Heptylphenol | 37c |
| H | H | C6H5 | p-Phenylphenol | 36d |
| H | H | OC6H5 | p-Phenoxyphenol | 36d |
 |
||||
Shortly thereafter, Ikeda and co-workers reported the polymerization of fluorine-containing phenols by HRP in a buffered mixture of water–organic solvent. This accomplishment was very important since fluoropolymers coatings are extensively used in industrial fields (e.g. Teflon™).38 Three monomers (Fig. 2) that have low oxidative reactivity were successfully polymerized besides reaction conditions such as organic solvent or buffer pH used were particularly investigated with 2,6-difluorophenol (2). The obtained polymer showed good water repellent property and mainly comprise 2,6-difluoro-1,4-oxyphenylene units, although when 3- or 4-fluorophenol monomers 3, 4 were used the polymer exhibited a mixture of phenylene and oxyphenylene units.
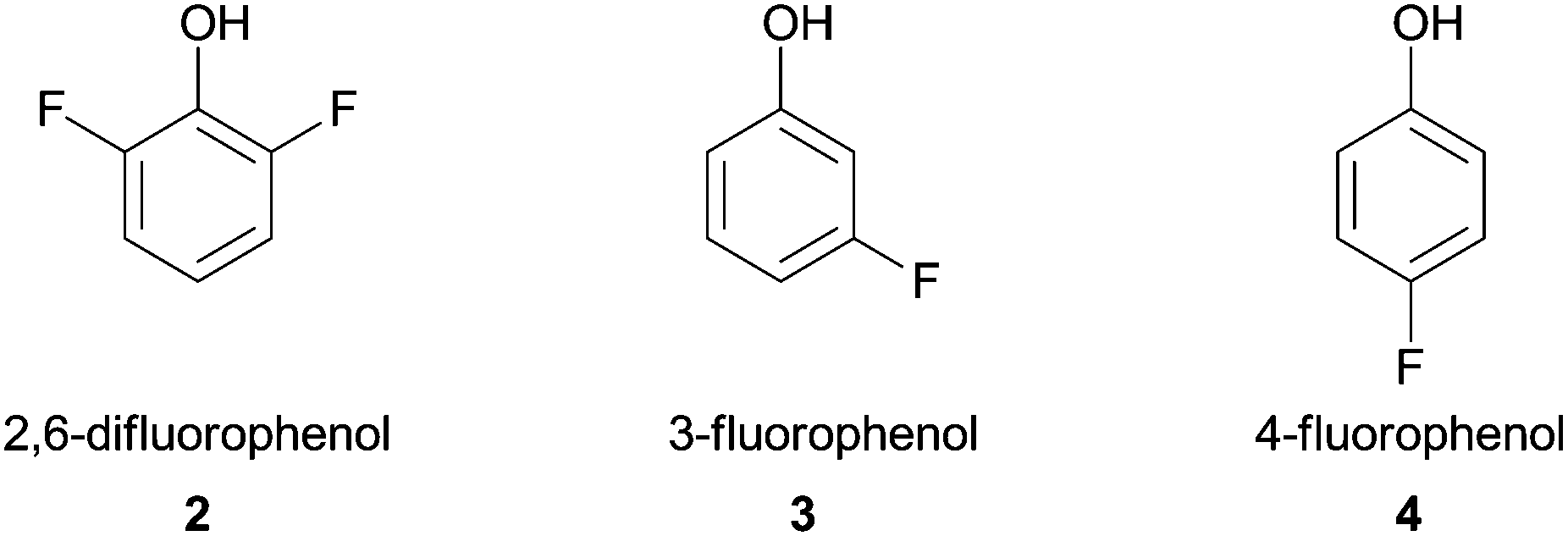 | ||
| Fig. 2 Fluorine-containing phenols polymerized by HRP. | ||
The kinetics of the phenol polymerization process was also studied, using high-performance liquid chromatography (HPLC), and it was verified that the reaction rate was higher when para- and meta-substituted phenols were used as substrates.39 There are important reaction conditions such as hydrogen peroxide (H2O2), the principal oxidant used in HRP reactions, temperature or buffer/organic solvent content that should be well-thought-out when planning an enzyme-catalysed polymerization.40 In fact, all these parameters greatly influence enzyme activity and consequently polymerization process. Traditionally polymerizations occur with harsh conditions to which enzymes do not resist, so the amount of H2O2 used is very important taking into account that its excess may inhibit the enzyme22e,41 as well as higher reaction temperatures decreases the polymer yield.40a One may conclude from the reported works that moderated temperatures and sequential addition of small amounts of H2O2 will lead to enhanced polymer yield.
One of the main concerns in green chemistry is to reduce waste generation that can be achieved, reducing and/or eliminating the use of organic solvents. The main idea is to develop environmental benign processes which do not deal with organic solvents; however phenols polymerization hardly proceeds in buffer medium.36a,42 The most standard solvent conditions are mixtures of organic solvents with buffer solutions. The researchers usually try to enhance the water content and one interesting example is the HRP catalysed polymerization of p-tert-butylphenol where the water content used point towards a modification of the polymeric structure.43 In the last decade, efforts have been made to develop methodologies that avoid organic solvents but although their claimed efficiency their applicability in industry has not been proved yet.
Zhang and co-workers studied the phenol polymerization by HRP in an aqueous micelle system using sodium dodecyl benzene sulfonate (SDBS) as surfactant.32 The addition of the surfactant greatly enhances the polymer yield and it could be obtained almost quantitatively. Furthermore high yields were maintained over a wide pH range (from 4 to 10) and the aqueous micelle system allowed that polymerization occurred in only 2 hours. The polymer exhibits limited solubility and high thermal stability. More recently these authors reported that sodium dodecyl sulfate (SDS) was more effective compared with SDBS.33 In fact, when polymerization was performed in phosphate buffer (pH = 7.0) the final conversion was less than 6%, while the addition of 0.15 g of SDS enhance it to higher than 90% which was higher than the observed in the previous study with the same amount of SDBS (less than 50%). The polymeric structure obtained is partly soluble in solvents as acetone, THF or DMF, being the polymer composed of a mixture of phenylene and oxyphenylene units in both cases.
Template polymerization where monomers units are organized by interactions with a template macromolecule is receiving special attention. Kim and co-workers performed the enzymatic polymerization of phenol in water, which proceeded regioselectively in the presence of poly(ethylene glycol) (PEG). High yields were achieved and the polymer precipitate as a complex with PEG.44 The results indicate that the presence of the PEG template improved the regioselectivity of the reaction since the phenylene unit content is higher than 90%. A great accomplishment of this work was that poly(m-phenylene) was obtained without the use of any organic solvent. In fact, polymers can be produced effectively from phenol and phenol derivatives using mixtures of organic solvents and buffer, but the polymerization in water is more difficult and the yields are low, so the above mentioned results proved to be a good improvement. The same authors also used poly(ethylene glycol) monododecyl ether (PEGMDE) as template in the polymerization of phenol by HRP in water. In this case the presence of this widely used non-ionic polymer surfactant also greatly improved the regioselectivity of the reaction since the phenylene unit content was close to 90%.45 Moreover PEG-poly(propylene glycol) (PPG)-PEG triblock copolymers (known as Pluronic) were also used in phenol polymerization and its addition prevented the polymer precipitation that can occur and hamper the polymer growing.46 This additive allowed a homogeneous polymerization yielding polymers with molecular weight higher than 106 Da instead of polymers with molecular weight of several thousand Da normally obtained in enzymatic polymerization of phenol. Furthermore, using Pluronic with high PEG content the regioselectivity obtained was improved leading to a polymer mainly containing phenylene units.
The above mentioned examples elucidate that the enzymatic polymerization produce polymers with a prevailing phenylene-structure, probably because they show high affinity to aqueous environments due to the free standings OH groups.
Peng and co-workers, knowing that carbon nanotubes (CNTs) are reported as a good support for enzymes immobilization and an outstanding material for polymer composites,47 used CNTs as templates in the regioselective synthesis of phenolic polymers.48 Their work allowed the synthesis of a polymer with nearly 90% of highly thermally stable oxyphenylene units, being this material the first example of an oxyphenylene structure-prevailing polymer produced enzymatically in water. Apparently the amount of CNTs used influence the polymer yield and regioselectivity.
Cyclodextrin derivatives (CD) are a series of cyclic oligosaccharides consisting of six (α), seven (β) or more D-glucose units connected by α(1→4) linkages. These structures were also successfully used as additives in HRP catalysed phenol derivatives polymerization. One of the advantages on the use of CDs is the accommodation of water-insoluble molecules inside their hydrophobic cavity which improves the solubility of such molecules. Thus, the use of organic solvents becomes unnecessary. Phenol itself was polymerized in a buffer solution in the presence of a catalytic amount of 2,6-di-O-methyl-α-cyclodextrin, producing a soluble polyphenol that contain a small amount of α-CD derivative.49 The same methodology, but with 2,6-di-O-methyl-β-cyclodextrin, was also efficient in the polymerization of m-substituted phenols in buffer with HRP.50 Although, in this case, a slight excess of β-CD relatively to the monomers was required for its solubilisation in the buffer, the β-CD derivative can be completely removed in the end of the reaction. CD derivatives were also used in the polymerization of other type of phenolic derivatives with HRP. From the several examples that can be found we single out the polymerization, in water, of hydrophobic bifunctional phenols such as N-(4-hydroxyphenyl)maleimide, 4′-hydroxymethacrylanilide and N-methacryloyl-11-aminoundecanoyl-4-hydroxyanilide-4-hydroxyphenylmaleimide,51 coniferyl alcohol, leading to dehydrogenative polymer with 8-O-4′-richer linkages,52 and in the oligomerization of ethyl 1-[((4-hydroxyphenyl)aminocarbonyl)]-2-vinylcyclopropane carboxylate, a para-functionalized phenol derivative.53
The recent interests in ionic liquids, which are viewed as an environmental friendly solvent alternative to the organic ones, stimulate researchers to use them in polymerization reactions with HRP. In fact, they may improve the solubility of phenolic monomers and provide a stable environment for the enzyme (among other promising characteristics).54 However there is still some controversy about its efficiency. For instance, Sgalla and co-workers pointed out that polymeric structures were not obtained when water insoluble phenolic substrates such as 1-naphthol, 4-phenylphenol and 2,5-di-tert-butylphenol were used in HRP reaction in 1-butyl-3-methylimidazolium tetrafluoroborate [BMIM][BF4]–water mixtures.55 Instead hydroxylated compounds and/or dimeric structures were obtained, and the authors justify the absence of polymeric structures as a consequence of the ionic liquid presence. Recently Zaragoza-Gasca and co-workers reported the use of [BMIM][BF4] to perform the polymerization of phenols in a mixture of ionic liquid and buffer.56 They obtained polymeric structures with molecular weights of 7000 kDa in 100% yield. Moreover, they also used the same ionic liquid for the synthesis of a photoconductive material, the poly(4-fluoro-2-methoxyphenol), showing that HRP enzymatic catalysis can produce polymeric structures useful for photoelectronic applications.57
A great advantage of enzymatic polymerization versus conventional methods is the high selectivity that can be achieved. From our literature survey several remarkable examples can be underlining mainly because when the used phenol derivatives present more than one polymerizable group the HRP can select one. The structural diversity obtained in this way is outstanding and new classes of highly reactive polymeric structures with a polymerizable group in the side chain can be obtained. For example, in the polymerization of 4-hydroxyphenethyl methacrylate (5) catalysed by HRP the phenolic moiety was chemoselectively polymerized, whereas with 2-phenylethyl methacrylate (6) the vinyl polymerization is favoured (Scheme 1).58
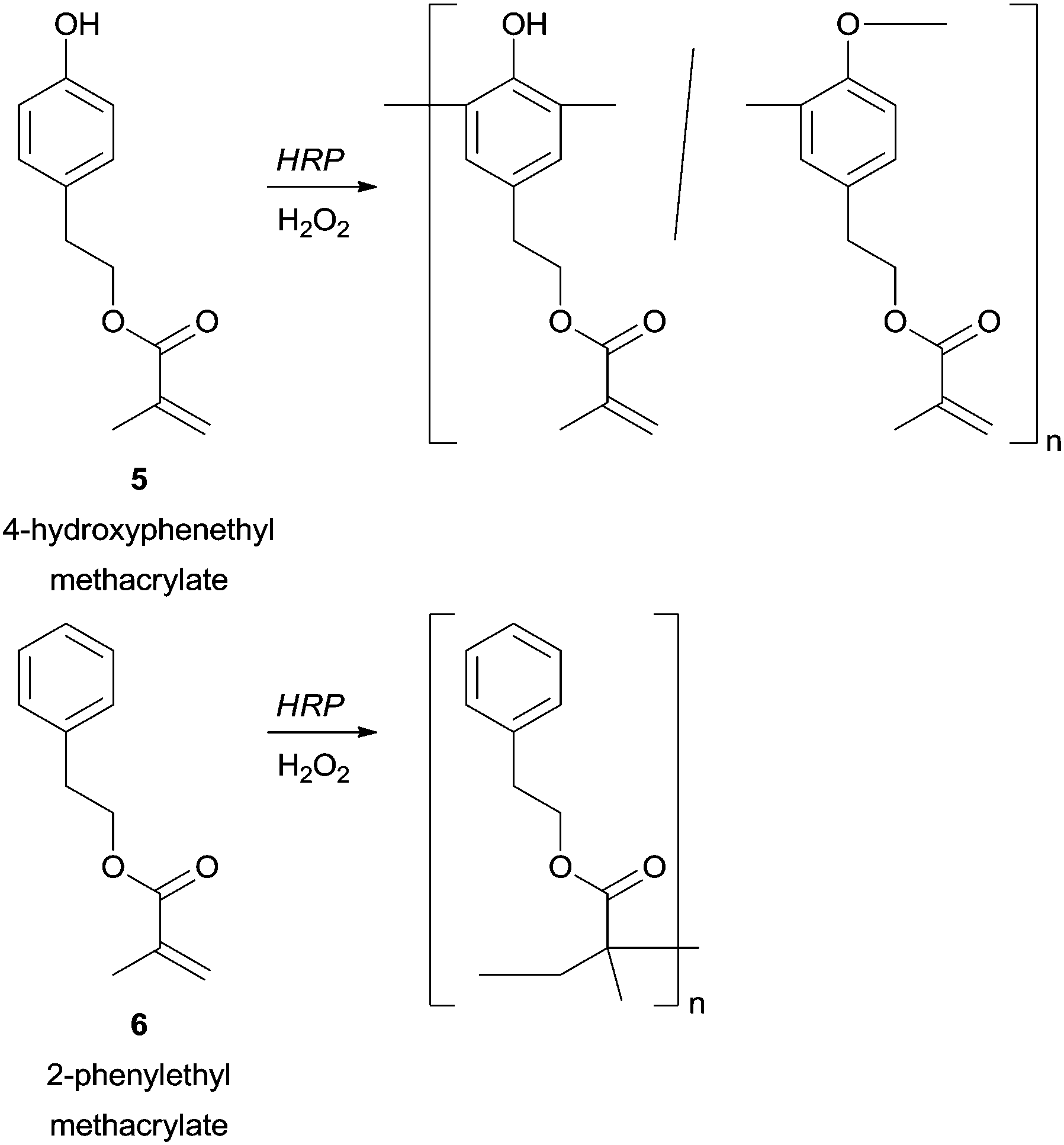 | ||
| Scheme 1 Chemoselective polymerization catalysed by HRP. | ||
Groups such as carboxylic or formyl can also be present in the phenol moiety and left untouched for further transformations in the polymer. For instance, in a mixture of methanol/phosphate buffer p-hydroxycinnamic acid (7) was successfully polymerized with HRP giving a polymer with carboxylic free groups and with the highest yield achieved using 60% methanol (Scheme 2).59
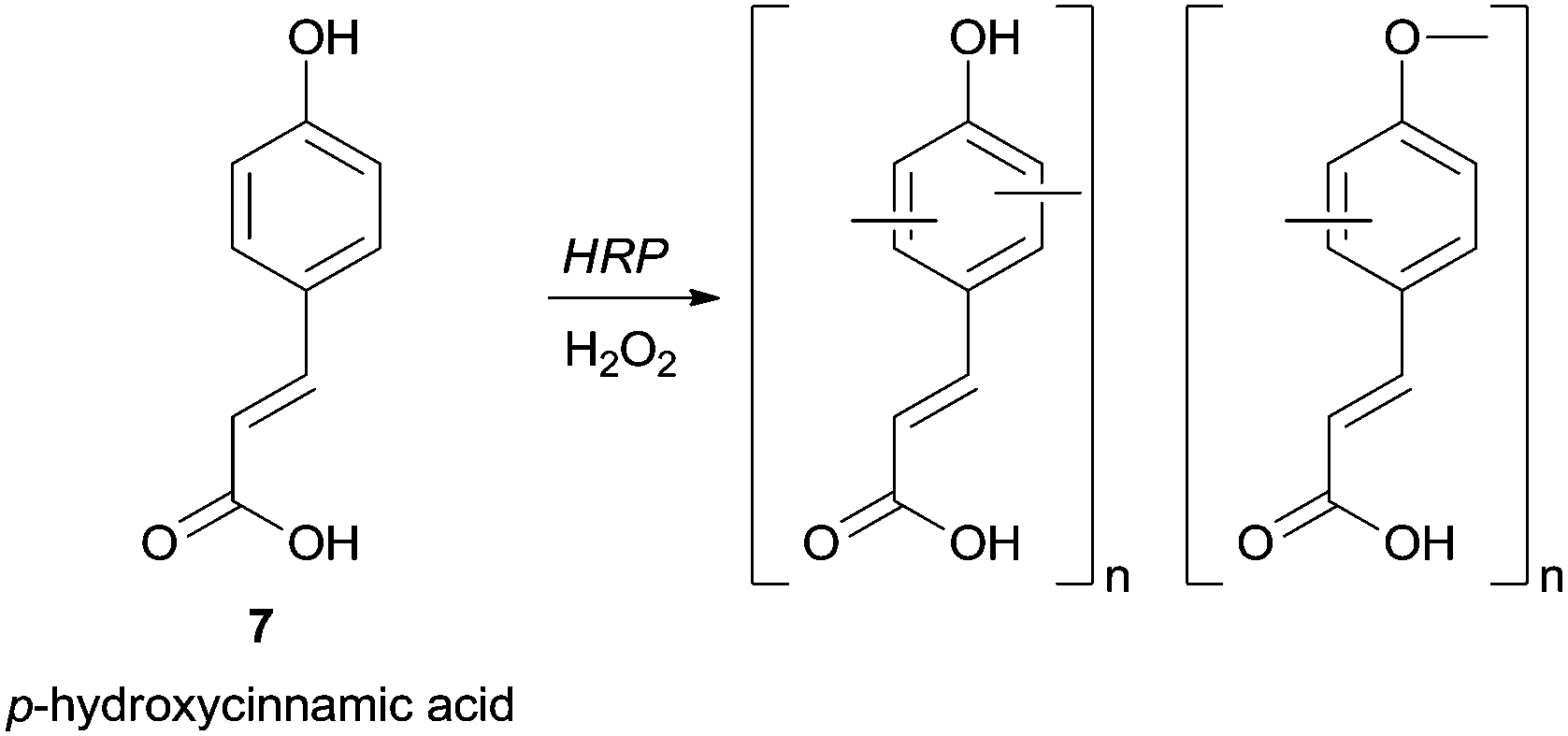 | ||
| Scheme 2 Chemoselective polymerization of p-hydroxycinnamic acid (7). | ||
Although the formyl group is very susceptible to be oxidised, polyhydroxybenzaldehydes could be polymerized in good yields by HRP.60 Interestingly with monohydroxybenzaldehydes as monomers the reaction does not occur, being the enhanced electron density of the aromatic rings due to more hydroxyl substitutions the trigger for the reaction. The aldehyde side groups in the polymer chains remain and no further oxidised to carboxylic acids. Nevertheless in some cases a previous protection was necessary. One of such examples is the copolymerization of p-hydroxybenzaldehyde (8) (HBA) and p-phenolsulfonate (9) with HRP/H2O2 system, being the formyl group protected via acetalization (Scheme 3).61 After the polymerization reaction, the final step is the formyl deprotection via acetal hydrolysis. The poly(p-hydroxybenzaldehyde-co-p-phenolsulfonate) obtained is a very interesting structure bearing a large conjugated system with active formyl and hydroxyl groups suggesting that can be used as agent for tanning leather.
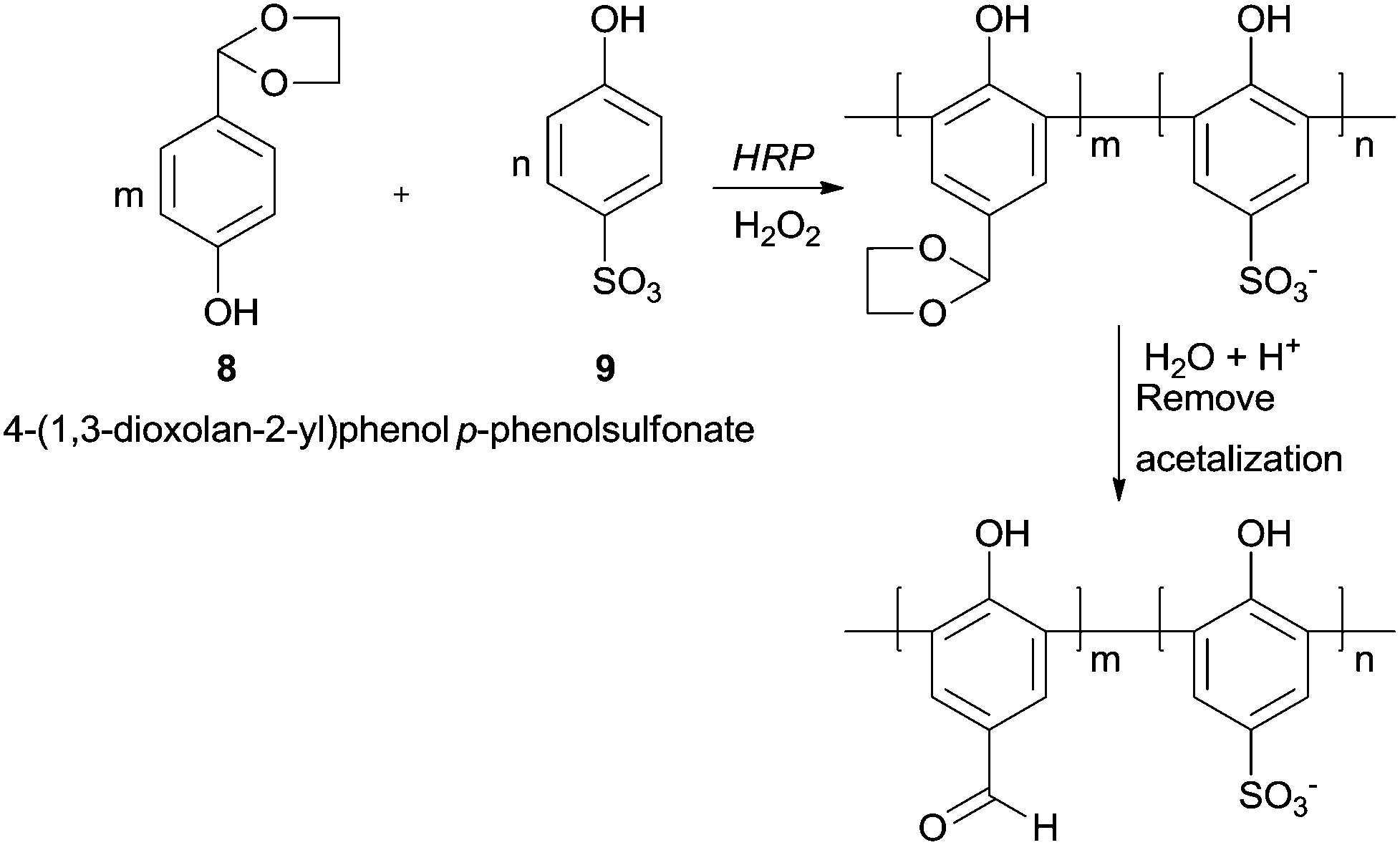 | ||
| Scheme 3 The formation of poly(p-hydroxybenzaldehyde-co-p-phenolsulfonate) by HRP with previous formyl group protection. | ||
The m-ethynylphenol (10) polymerization, studied by Tonami and co-workers, is another extraordinary example of HRP selectivity.34b Let is look carefully to the reported results (Scheme 4). m-Ethynylphenol (10) possess two reactive sites, the acetylene and the phenol moieties, and in addition to HRP catalysed reaction a conventional copper/amine catalyst was tested. The phenolic moiety was polymerized with HRP while the ethynyl group remains unaffected in the side chain. On the other hand, the conventional catalyst produced a diacetylene derivative via dimerization by ethynyl group so the authors demonstrated that the enzyme induced chemoselective polymerization.
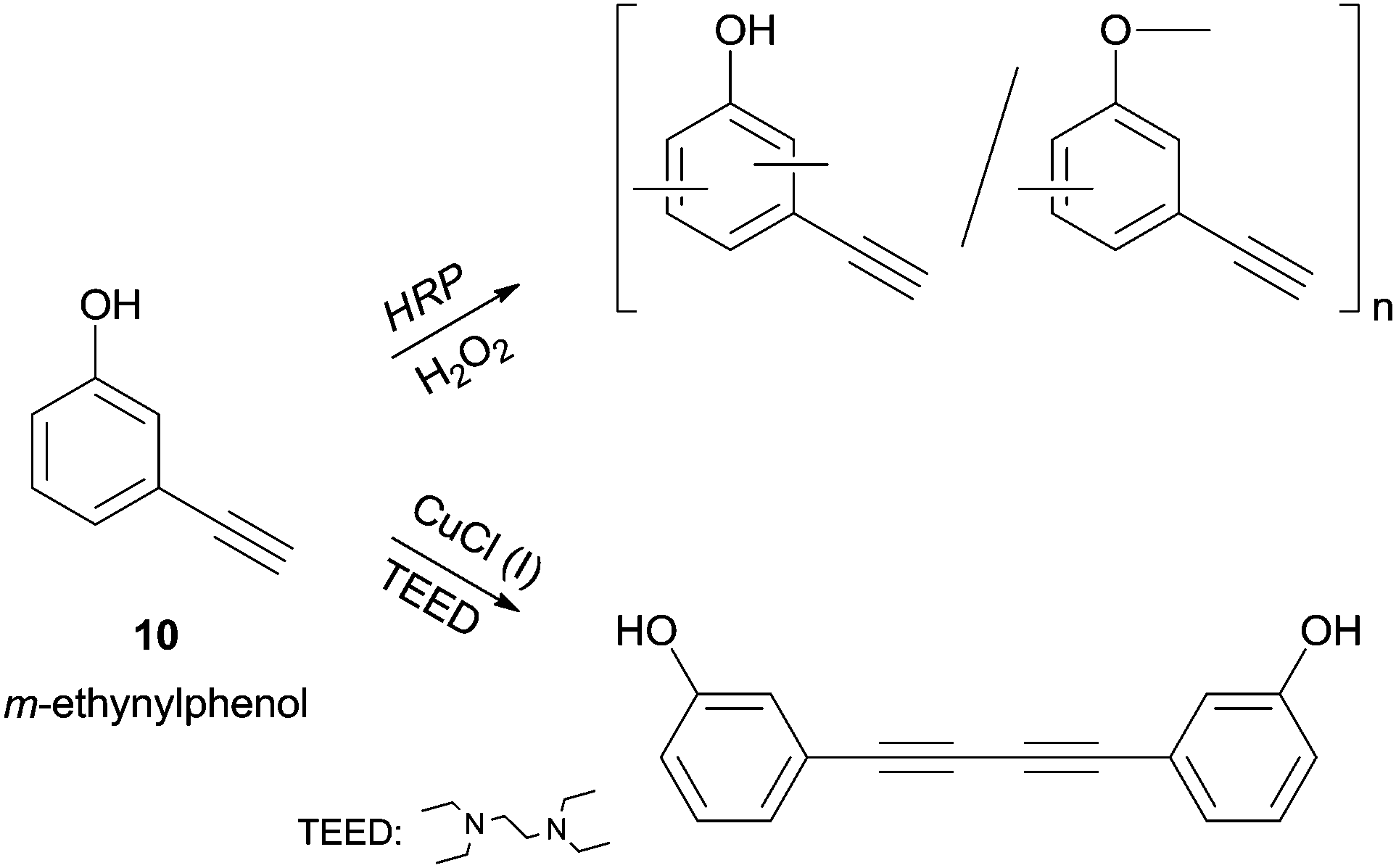 | ||
| Scheme 4 Polymerization of m-ethynylphenol (10) by HRP as opposed to dimerization by conventional catalyst. | ||
It is true that phenol and phenol derivatives have been clearly the most studied substrates regarding enzymatic polymerization by HRP over the years. However, other types of compounds have been tested as monomers to obtain polymeric structures in an environmental benign way with HRP and aniline have also received particular attention. Interestingly, the different behaviour of these two types of substrates, phenols and anilines, during HRP reaction has already been the subject of investigation.62 In fact, the synthesis of polyaniline is really attractive due to its wide range of electrical, electrochemical and optical properties allied with their good stability. However, even though that the aniline and aniline derivatives polymerization is important and interesting its discussion must be covered elsewhere. On the other hand, we could not skip examples of polymerizations of phenols bearing amino groups. An example is the work of Reihmann and Ritter where the commercially available and electron rich 4-aminophenol (11) was used as monomer to build up a redox active phenol polymeric structure using HRP as catalyst.63 The reaction of HRP/H2O2 with unprotected 4-aminophenol (11) led mainly to the formation of 1,4-benzoquinone-monoimine 12 whereas the reaction with the protected monomer made possible the formation of phenol polymers (Scheme 5). The authors used 4-nitrobenzaldeyde in the protection step being formed 4-(4-nitrobenzylideneamino)phenol (13) and the corresponding acid in the posterior deprotection final step. Another interesting result that was useful to understand the polymerization process was also reported. If the reaction is ended up with catalase at an early stage, dimers of 4-(4-nitrobenzylidenoamino)phenol (14) were obtained and the existence of mainly 2,2′-dihydroxybiphenyl derivatives suggested that the polymer structure was composed by 1,3-phenylene linkages. Moreover, it was showed that mainly dimers are formed in the beginning of the polymerization process, which then undergoes radical transfer and recombination processes.
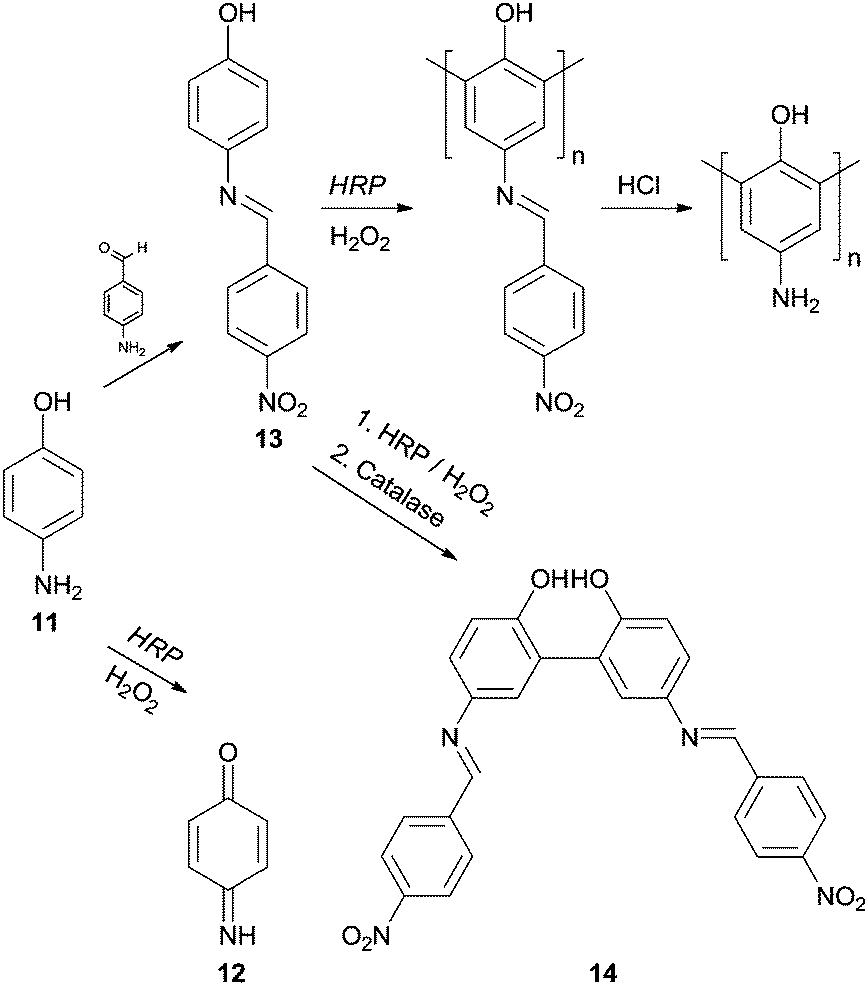 | ||
| Scheme 5 Pathways of transformation of 4-aminophenol (11) via protection/deprotection of monomer. | ||
A research area that has received particular attention is the synthesis in confined volumes at a micro scale. In this field, microcapsules obtained in layer-by-layer assemblies of oppositely charged polyelectrolytes, proteins and nanoparticles allow the creation of thin multilayer films with nanometer thickness and enable mimicking processes that occur in living organelles. Ghan and co-workers performed polymer synthesis within layer-by-layer polyelectrolyte microcapsules with HRP based on selective permeability of the capsule walls.64 In fact, they are penetrable for monomers but HRP and polymeric chains cannot leave capsules interior due to the high molecular weight. Fluorescent and easily detectable polymeric products were obtained being 4-(2-aminoethyl)phenol (15) (tyramine) hydrochloride used as monomer (Scheme 6). Thus, the possibility of synthesize functional materials, such as luminescent polymers, in the microcapsule becomes attractive. In these cases, the template-soluble cores used influence the capsule diameter which could be in the range of 100 nm to tens of microns.
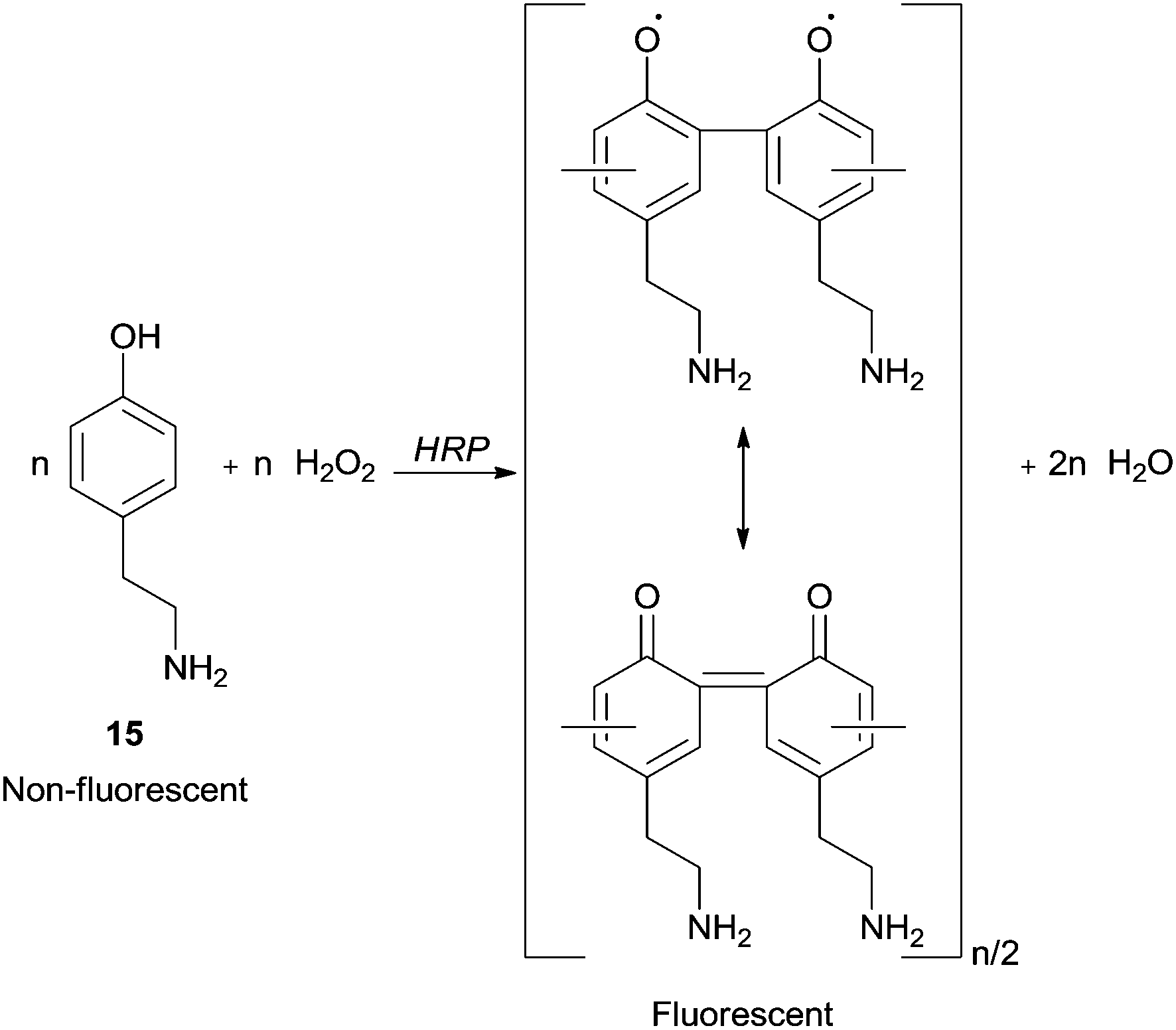 | ||
| Scheme 6 Formation of fluorescent polymeric structure by HRP reaction within microcapsule. | ||
Recently, it was reported the use of HRP/H2O2 to synthesize graft copolymers of degraded starch 16 with p-hydroxybenzoic acid65 (17) or with resorcinol66 (18) (Scheme 7). The obtained polymers showed excellent tanning properties, constituting an environmentally profitable process to leather tannage synthesis and a replacement for toxic chrome and aldehyde tannage.
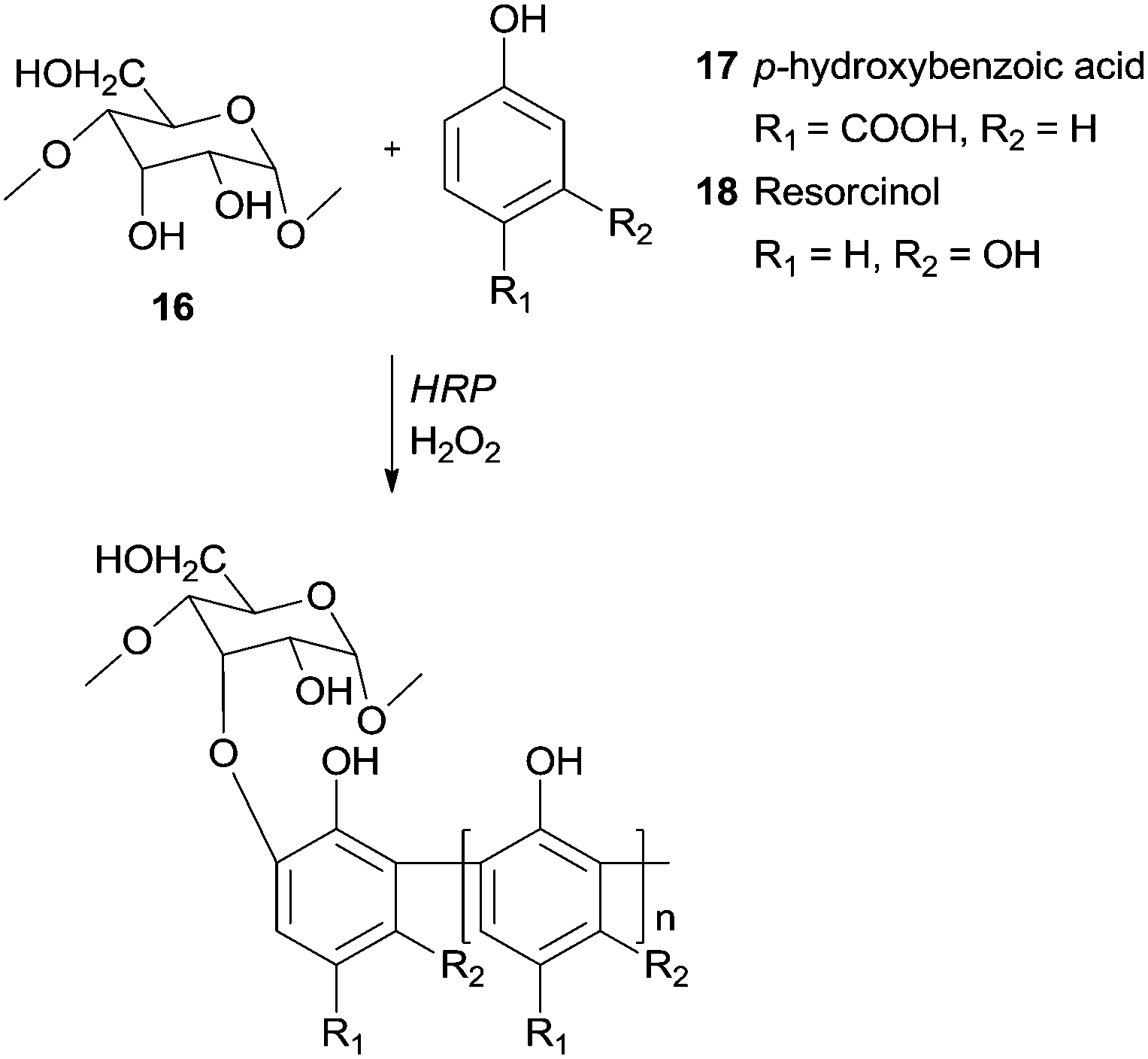 | ||
| Scheme 7 Graft copolymers of degraded starch 16 with p-hydroxybenzoic acid (17) and with resorcinol (18). | ||
More than four decades ago the HRP effect on the transformations of polyphenolic compounds was investigated, mainly to understand its role in biosynthesis.67 As far as we are aware one of the first examples of polymerizations studies started with chalcone derivatives but was not completely successful.68 From the several derivatives used, including a complex aminochalcone/2,6-dimethyl-β-cyclodextrin, the polymerization reaction occurred with only two aminochalcones and proceeds slowly in both cases. The resulting products had molecular weights in the range of 3000–3600 Da after four days of reaction. A few years later more interesting results were reported and some insights arose from that work.69 The authors found that the polymerization of some isoflavonoids and their precursors in aqueous media with the catalytic system HRP/H2O2 did not occur due to the weak solubility of the monomers. In fact, only daidzein derivatives with a 4′-hydroxyl group were successfully polymerized (Scheme 8). However if a methoxy or a nitro group or a hydrogen atom was present instead at this position the reaction did not occur. Daidzein (19), 5,6,4′-trihydroxyisoflavone (20), quercetin (23), rutin (24) and catechin (25) were polymerized through radical polyrecombination being obtained polymeric structures with molecular weight of 4000–12000 Da. It is interesting the fact that the polymerization reaction occurs through the electron-rich ring B (Scheme 8). The 5,6,4′-trihydroxyisoflavone (20) is a good antioxidant due to the presence of a catechol moiety in ring A that makes it very reactive towards free radicals. The yield of its polymerisation was low (30%), however, it is an interesting achievement the formation, by bio-catalysis, of a polymer composed by monomers sensitive to radicals. Low-molecular-weight products could also be obtained during the reaction since primary free radicals might recombine to oligomers or produce oxidation products. Factors as the low concentration of the substrate or reduced enzyme activity could lead to the oxidation of the free radicals due to the reduced probability of recombination. The pathway selected, polyrecombination or oxidation, is then defined by the competing kinetics of the processes.
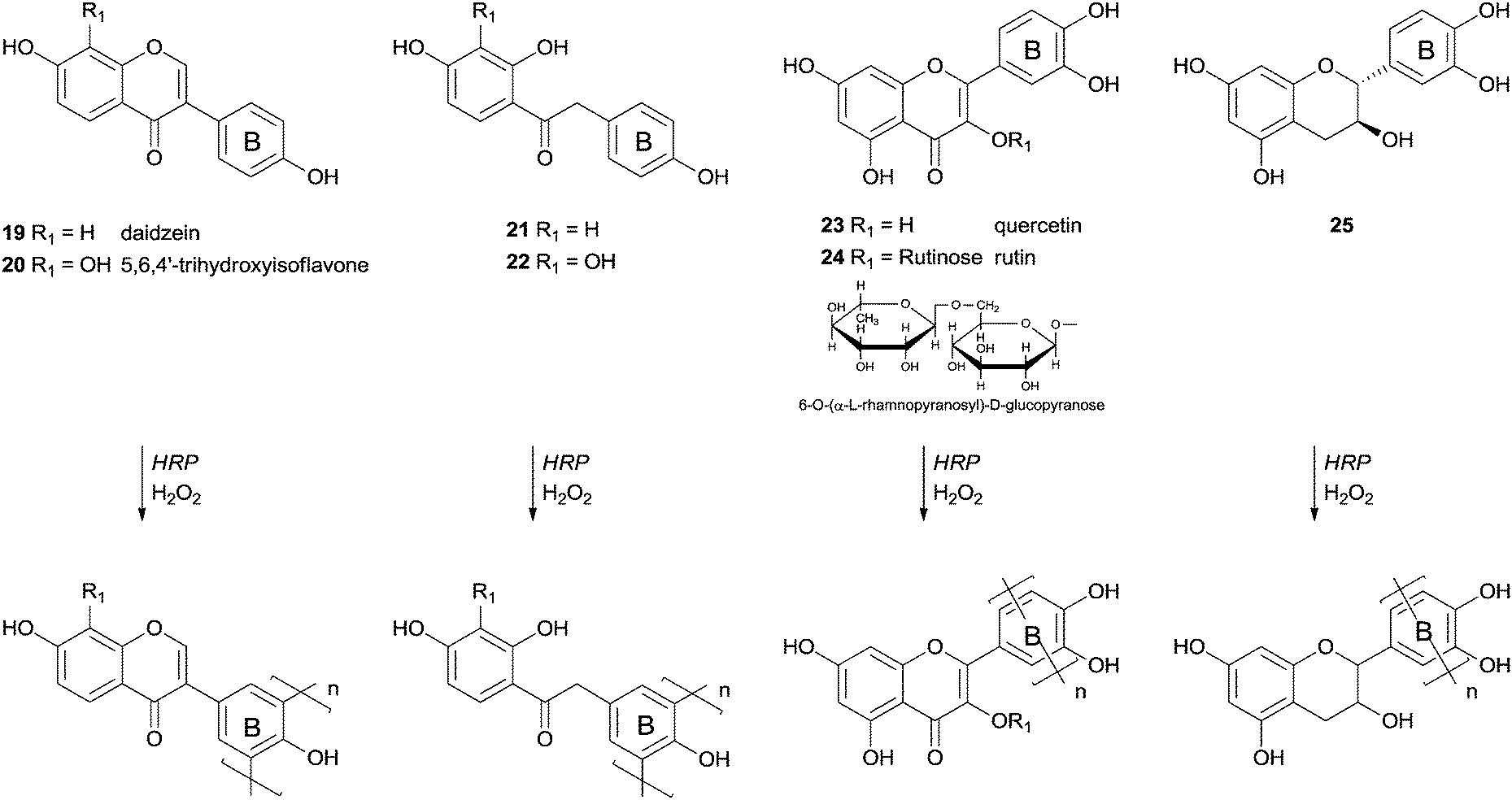 | ||
| Scheme 8 Polymerization of phenolic compounds by HRP. | ||
Quercetin (23) was used as a monomer in polymerization studies with HRP by other authors that were able to polymerize this compound in water–ethanol mixtures and obtain, via mild conditions, a soluble polymer (Scheme 9).70 The content of flavonoids and other polyphenols in food tend to decline owing thermal processing, as boiling or frying, but the authors claimed that the polymeric structures obtained are thermally stables. Therefore they can be used as potent antioxidants in the highly regulated food industry even because starting materials, the solvents and catalyst used are biocompatible. FTIR analysis showed that polyquercetin is highly hygroscopic and that a significant amount (75%) remains after polymer heating at 600 °C. This fact must be due to the formation of new catechol–catechol bonds in the polymer's backbone. Contrary to some other polyphenolic compounds synthesized, rotation of the bond C2–C1′, after polymerization, breaks the conjugation of the quercetin and the polymer does not contain considerable amounts of conjugated quercetin moieties. Although the authors propose the structure of the polymer presented in Scheme 9, they believed that it is very complex requiring further purification and characterization. They also reported that the enzyme immobilization would be a good process improvement in view of large scale production of processable phenolic resins based on natural monomers.
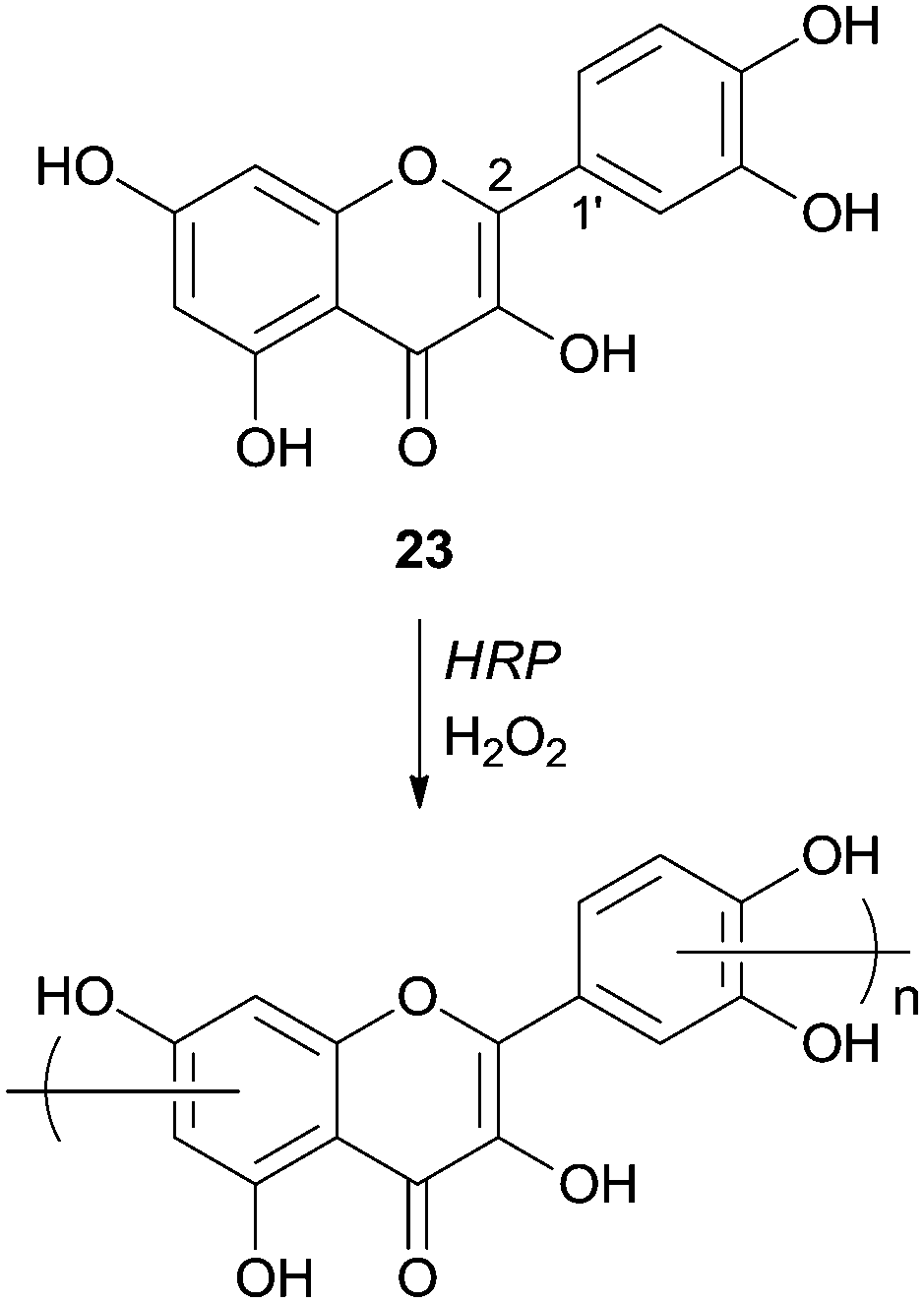 | ||
| Scheme 9 Polymerization of quercetin by HRP. | ||
4. Enzymatic coupling
Back to the 1980s and 1990s HRP was used in coupling reactions and several interesting compounds were produced. Moreover some of them showed important biological activities. We envisioned focusing our analysis on recent examples that illustrate the use of HRP and its green advantages in coupling reactions. However the studies on the oxidative coupling using this enzyme remounts to 1970s when Gochman and Schmitz reported the coupling of 3-methyl-2-benzothiazolone (MBTH) and N,N-dimethylaniline to form a water-soluble, stable and intensely coloured indamine dye 26 (Fig. 3).71 The enzymatic coupling reaction involving HRP was particularly studied in the last two decades of the last century and the reports in the begin of the 21st indicates a nonstop interest. Select examples from the huge amount of interesting work found in literature is a hard job and the choices herein presented were mainly done taking into account that they involve phenolic compounds and allowed the synthesis of exquisite compounds that can stimulate researchers to transformations involving biocatalysis. | ||
| Fig. 3 Examples of structures involved in coupling reactions. | ||
From the earliest examples one can highlight the HRP/H2O2 catalytic system used in the synthesis of guaiacol dimeric 27 and trimeric 28 anti-microbial structures,72 new spirodiastereomers 29, 30 obtained in the dimerization of (E)-methylsinapate73 and also its coupling with 1-(4-hydroxy-3,5-dimethoxyphenyl)ethanol obtaining a cyclohexadienone spiro compound 31 as main product.74 Another example is the catharanthine and vidoline coupling75 to form α-3′,4′-anhydrovinblastine 32 (Fig. 3), an important precursor of known anticancer drugs.75a
Quite interesting results were found when the HRP/H2O2 system catalysed reaction with chlorophenols.76 When 2,4- or 2,6-dichlorophenols were tested, dimeric structures were generated being followed by its subsequent oxidation to quinone derivatives.76a,b Furthermore it was found a pH-dependency related to the reaction rate. However, the dimeric structures were not always obtained as proved by the reaction with 2,4,6-trichlorophenol.76c In fact, the substitution of a chlorine atom with an oxygen atom occurred and the respective p-quinone derivative was formed instead.
Some success has been achieved with coupling reactions of cinnamic acid derivatives catalysed by HRP. One example is the use of chiral ferulic acid amides.77 The coupling reaction of ferulic acid amides with ethyl S-alaninate group as chiral auxiliary in dioxane–aqueous buffer pH 3 allowed the synthesis of a diastereomeric mixture of phenylcoumaran-type structures (β-5 dimer) in 70% yield. Even more important the chromatographic separation and hydrolysis showed the achievement of a significant enantiomeric excess (ee 65%) being this the first example where stereocontrol was observed in enzymatic coupling reactions of phenylpropenoidic phenols.77b Later on cross-coupling reactions of equimolecular mixtures of ferulic and sinapic acids amides, with a R-methyl benzyl amine group as chiral auxiliary, gave diastereomeric excess of the new dihydrobenzofuran bis(R-methylbenzylamide) (33). However, β-5 benzofuran 34 from dimerization of ferulic methyl benzyl amide and β-β-aryltetralin 35 from the dimerization of sinapic methyl benzyl amide were also detectable products (Fig. 4).77a The observed regioselectivity have not been elucidated yet which is still a challenge for researchers.
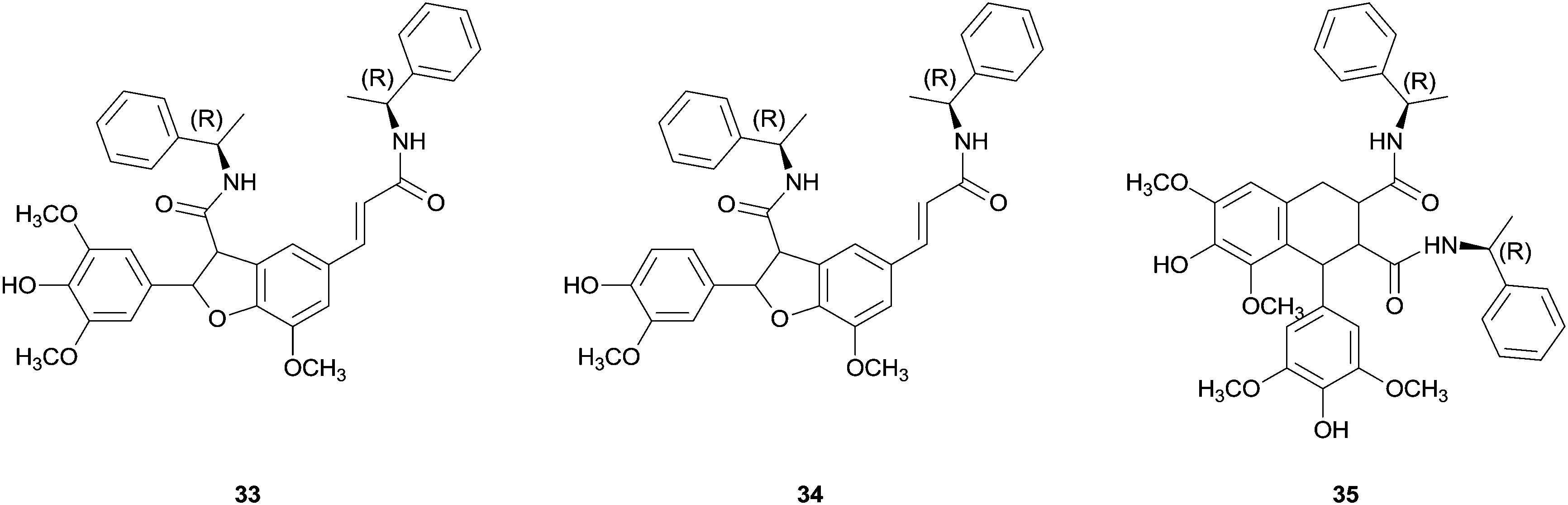 | ||
| Fig. 4 Products of the cross-coupling reactions of ferulic and sinapic acids methyl benzyl amides. | ||
In the same report, the synthesis of new dihydrobenzofuran lignans with a phenylcoumaran skeleton via cross-coupling reaction of methyl esters of substituted hydroxycinnamic acids (Fig. 5) was also reported. The authors recall the possibility that these new unnatural heterodimers may offer better bioactivity than the natural analogues. In addition, this kind of study is biologically important to understand the preponderance of cross-coupling reactions in lignification process being the oxidation potential and the radical reactivity important factors regarding this field.78
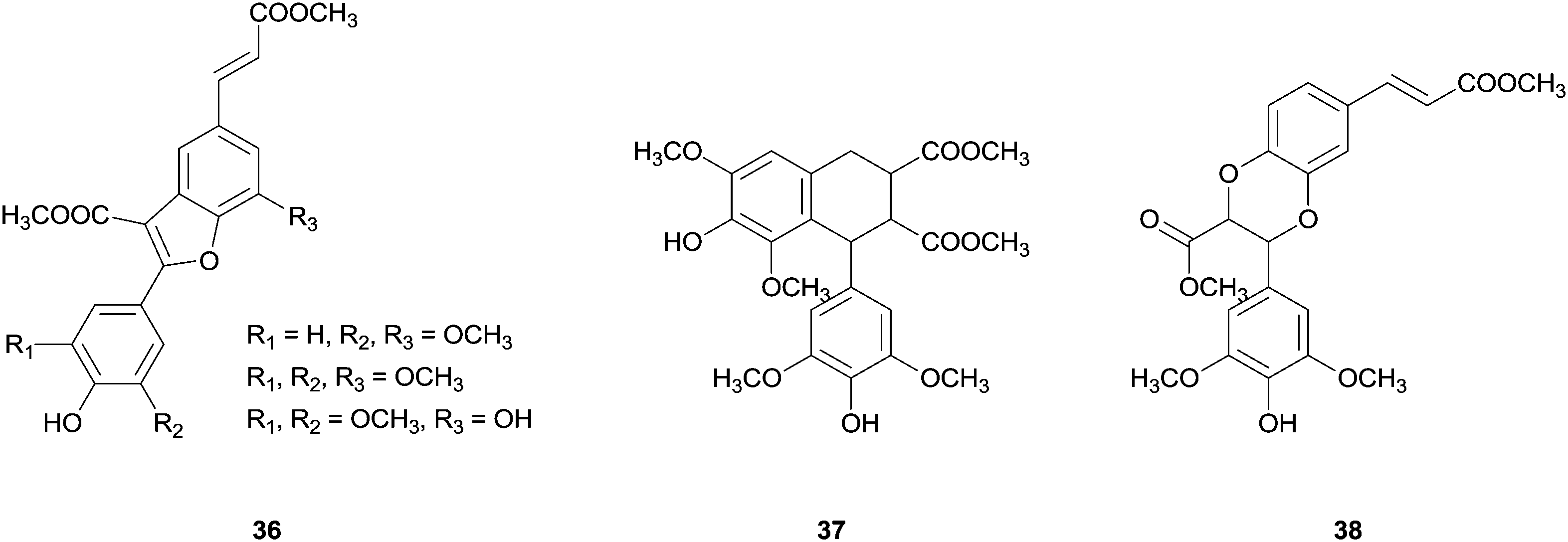 | ||
| Fig. 5 New dihydrobenzofuran lignans obtained from the cross-coupling reactions of methyl ester of substituted hydroxycinnamic acids. | ||
Other examples of fruitful coupling reactions using ferulic acid derivatives can be found. For instance Oppolzer's camphor sultam and Evan's 2-oxazolidinone derivatives of ferulic acid were used in coupling reactions and dimeric benzofuran neolignan structures were enantioselectively obtained.79 In another report, HRP-induced coupling reactions of equimolecular mixtures of substituted hydroxycinnamic acid suberin precursors (ferulic-, caffeic-, coumaric- and sinapic acid) showed that only two mixtures were found to undergo cross-coupling reactions.80 In fact, caffeic acid reacted with ferulic and also with sinapic acid leading to heterodimers. On the other hand, with the other mixtures, one of the substrates dimerize preferentially being obtained products that predominantly had β-β′-γ-lactone and β-5 benzofuran molecular frameworks. Recently HRP allowed the aryl–aryl coupling in a key step of the first total synthesis of the naturally occurring (5-5′)/(8′-O-4′′)-dehydrotrimer of ferulic acid.81
In another fascinating field of application HRP showed the ability to catalyse the generation of the in situ gellable sugar beet pectin (SBP) due to the coupling of feruloyl groups 39 and being the SBP aqueous solutions gelled within one min of reaction (Scheme 10).82 An interesting fact is that this mixture was successfully gelled subcutaneously in the presence of the enzyme and H2O2. Furthermore, and vital to biomedical fields induced necrosis was not noticed in the surrounding tissue after one week of application.
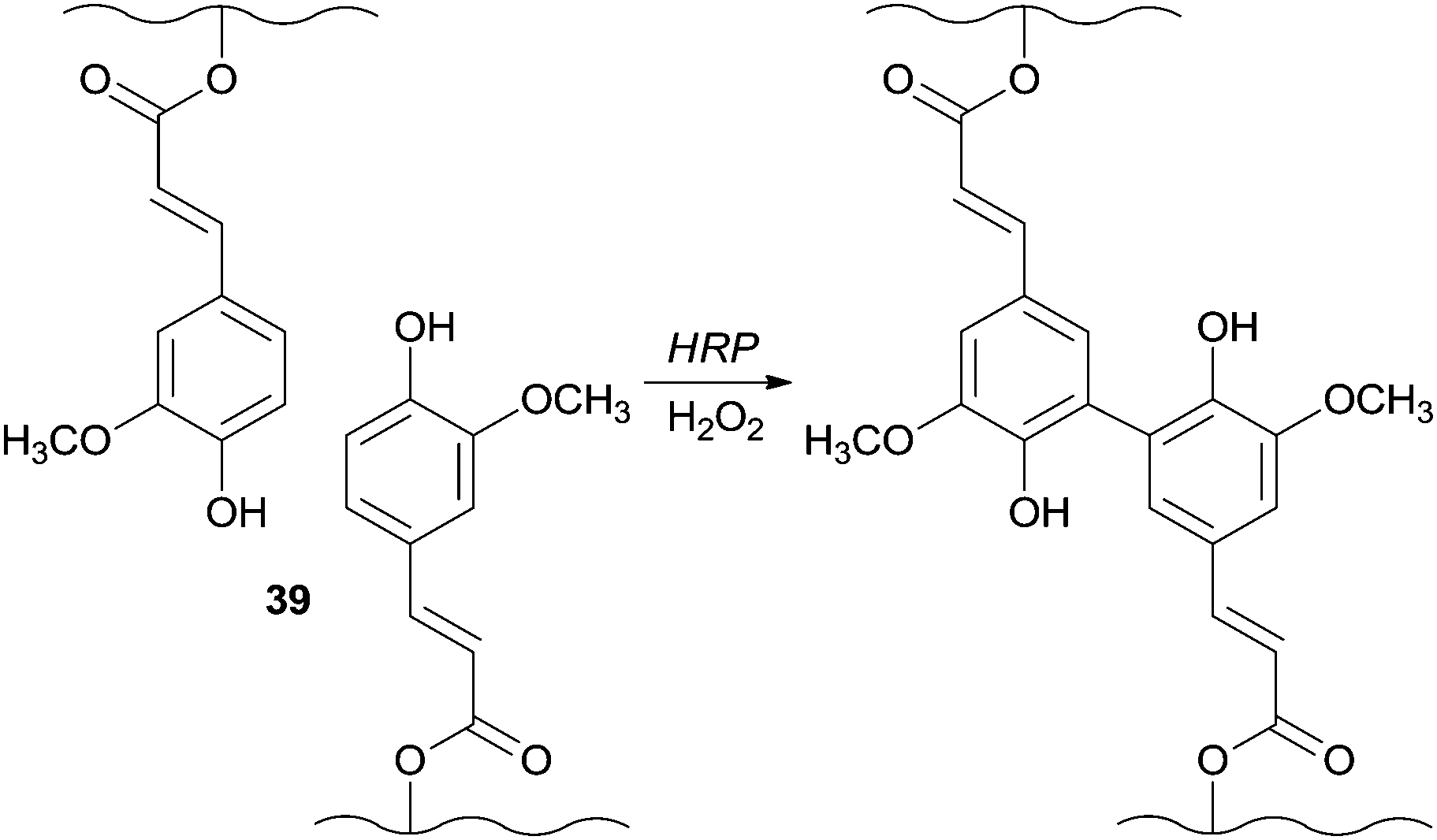 | ||
| Scheme 10 Formation of in situ gellable sugar beet pectin (SBP). | ||
The kinetics and rheology of this reaction was also studied recently.83 In fact, current reports elucidate that HRP/H2O2 system can be very helpful in medicine,84 as a recent review shows that several hydrogels used for tissue engineering were obtained exclusively when enzymes were used.85
Spectrofluorimetry and differential spectrophotometry were used by Tang and co-workers to study the HRP/H2O2 catalysed oxidation of tyrosine (Tyr) (40) in water and alcohol (methanol, ethanol, 1-propanol and isopropanol)–water mixtures (Scheme 11).86 They observed that the fluorescence intensity of the product weakened in the alcohol–water mixture compared with the reaction performed only in water. In fact, when a small amount of alcohol was added the enzyme activity was probably enhanced and probably had a sensitization effect on the fluorescence of Tyr dimer, but a greater quantity of alcohol reduced the fluorescence intensity. The authors suggested that probably the yield of Tyr dimer decreased and non-fluorescent polymeric structures were formed, suggesting that the addition of alcohols must change the HRP conformation leading to polymerization, although the quenching of the Tyr dimer by the alcohols was also pointed out as hypothesis.
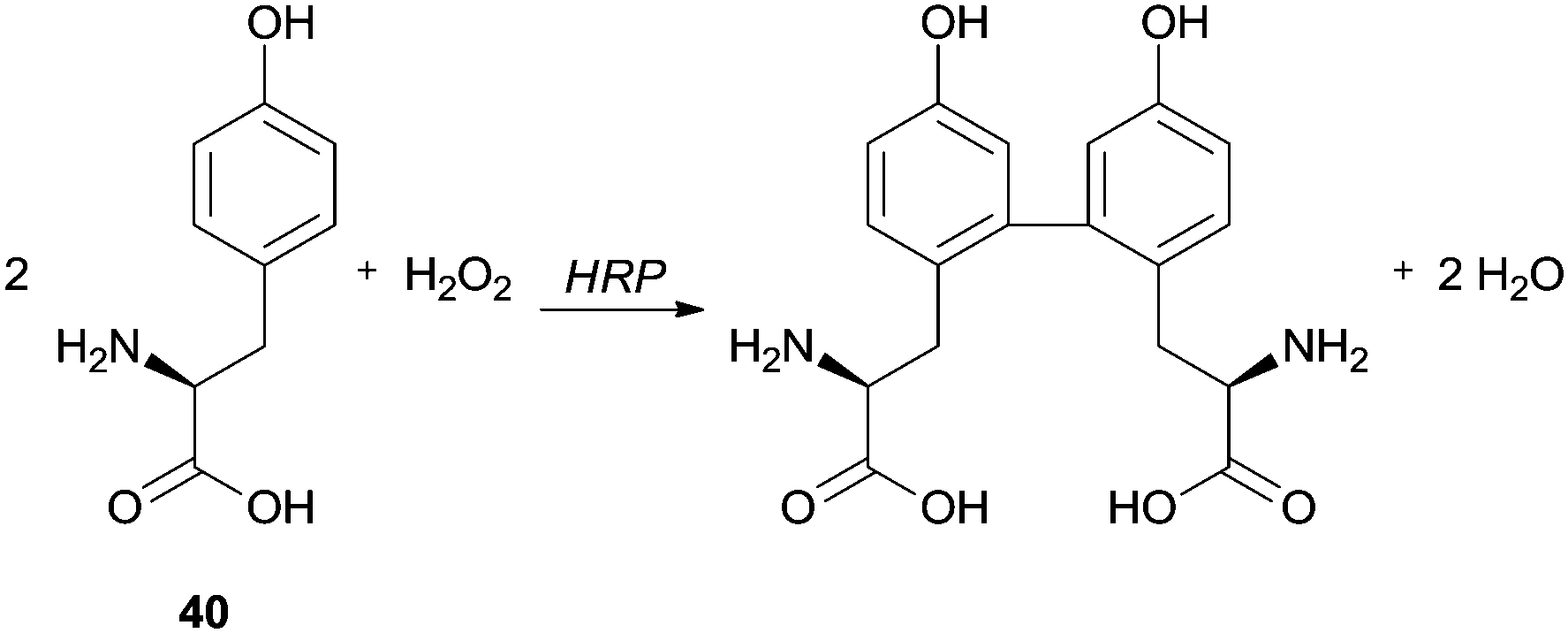 | ||
| Scheme 11 HRP/H2O2 oxidation of tyrosine (Tyr) 40. | ||
Moreover it was reported that HRP-mediated the linkage of tyrosine-containing peptide Gly-Tyr-Gly (GYG) with 3-(4-hydroxyphenyl)propionate saccharides (Scheme 12).87
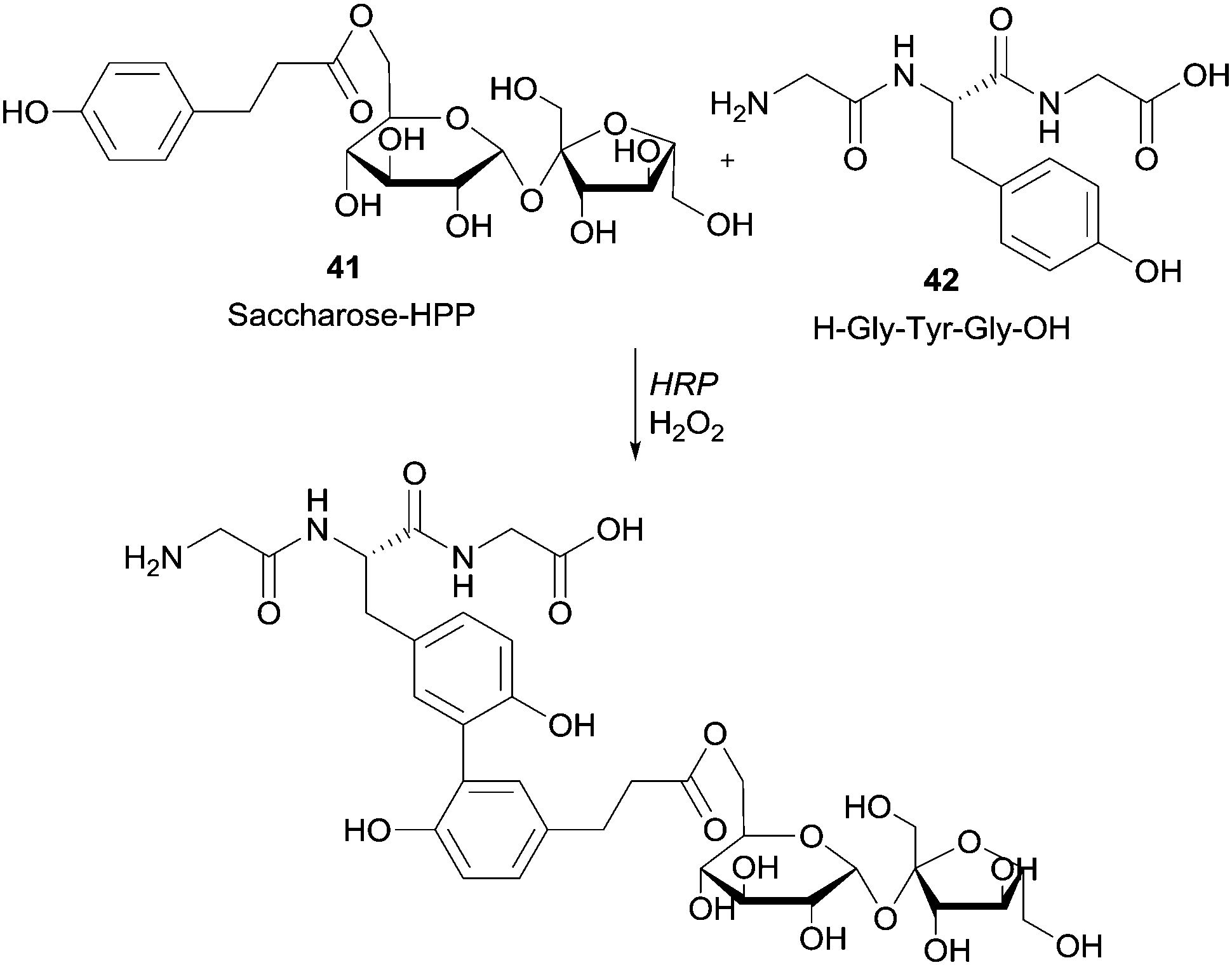 | ||
| Scheme 12 Coupling of hydroxyarylated saccharose 41 to the tripeptide GYG 42 by HRP. | ||
Previously, the functional phenolic moiety was connected to oligossacharides and glycosidic polyols, so several di- and trisaccharides were hydroxyarylated. The authors used the representation shown in Scheme 12 to illustrate the linkage of hydroxyarylated saccharose 41 to the tripeptide GYG 42, drawing attention to the fact that there are various possibilities for the position of the link. The successfully enzymatic linking between the tyrosine-containing peptide and the saccharose mono-ester is regarded as a model for enzymatic protein glycation and seen as an opportunity for the development of products with new functional characteristics. The same aforementioned peptide GYG was cross-linked to ferulic acid.88 The major product was formed through the dehydrogenative linkage of two ferulic acid molecules to a single peptide, but also to peptide oligomers, ranging from dimers to pentamers. Furthermore, it was also observed that mono- and dimers of the peptide were linked to only one molecule of ferulic acid. However, in another report the same authors showed that quite different cross-linked products could be formed with the same start materials.89 Thus, they believed that factors such as substrate concentration greatly influence the course of the reaction. Anyway, this hetero-coupling explain the protein–carbohydrate complexes present in plant cell walls, as well as the incorporation of ferulic acid and other hydroxycinnamic acid derivatives into lignin and suberin tissues. We will not extend on what concerns the use of HRP in protein cross-linking and protein–protein heteroconjugation. However, it should be noted that new structures with different functions due to the combinations of divergent protein properties could be achieved. These novel conjugates may have a huge diversity of interesting applications.90 Regarding polysaccharides structures several reports describe the use of HRP in the study of cross-linking reactions involving namely arabinoxylans from wheat flour and wheat bran, pentosans, corn-bras hemicellulose or beet pectin extracts.91 Another type of saccharide, the 4′-hydroxyphenyl-β-D-glucoside (arbutin, Arb) (43) was coupled by HRP with 2,5-dihydroxybenzoic acid sodium salt (gentisate, GA) (44) and a new product with a novel aglycone structure was obtained (Scheme 13).92 The coupling reaction is followed by spontaneous intramolecular esterification between the phenolic hydroxyl group of Arb 43 and the carboxyl group of GA 44. The obtained product showed higher antioxidation activity and competitive inhibition against mushroom tyrosinase than Arb, but lower inhibition against melanoma tyrosinase. This kind of reaction profile provides efficient and specific modification of aglycone structures of phenolic glycosides without the need of protection and deprotection steps beyond that can be used to produce new building blocks for cosmetics and pharmaceuticals.
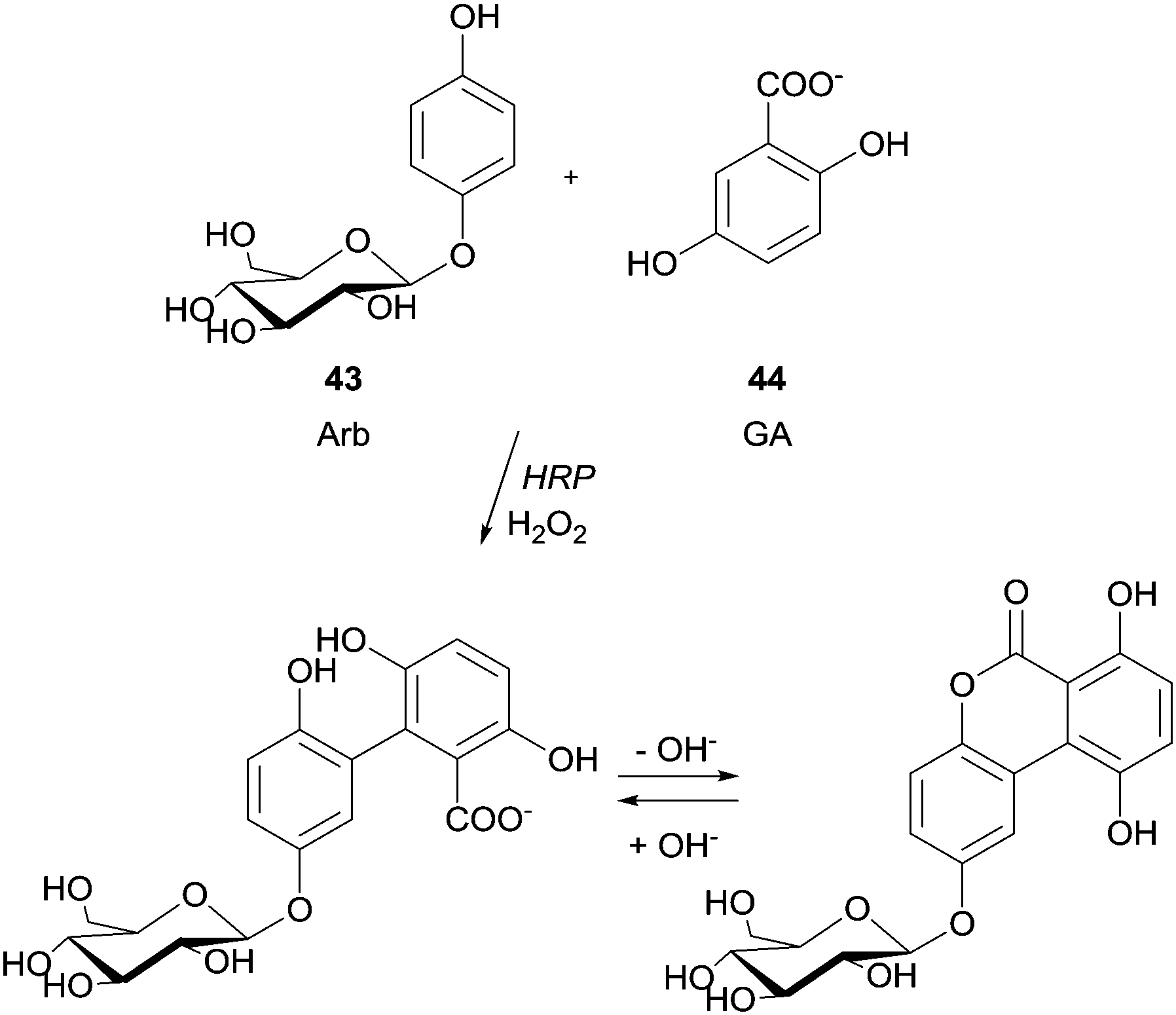 | ||
| Scheme 13 Coupling of 4′-hydroxyphenyl-β-D-glucoside (arbutin, Arb) (43) with 2,5-dihydroxybenzoic acid sodium salt (gentisate, GA) (44). | ||
Resveratrol (trans-3,5,4′-trihydroxystilbene) (45) is an abundant phytochemical occurring in the grape skin that has emerged great interest and already generated a wide amount of scientific reports due to its in vitro and in vivo antioxidant capabilities. In the mid-1990s, trans-resveratrol (45) was identified as one of the possible factors responsible for the French Paradox concept.93 Naturally its biotransformation using HRP and hydrogen peroxide was studied and trans-δ-viniferin (46), a resveratrol-trans-dehydrodimer was obtained as the major product (Scheme 14).94 Other studies can be found where this important polyphenolic compound was used as HRP substrate and, for instance, its dimeric structure (+)-ε-viniferin was also tested.95 Also the formation of (+)-davidiol A was achieved when HRP promote the oxidative dimerization of trans-resveratrol (45) and trans-ε-viniferin.96
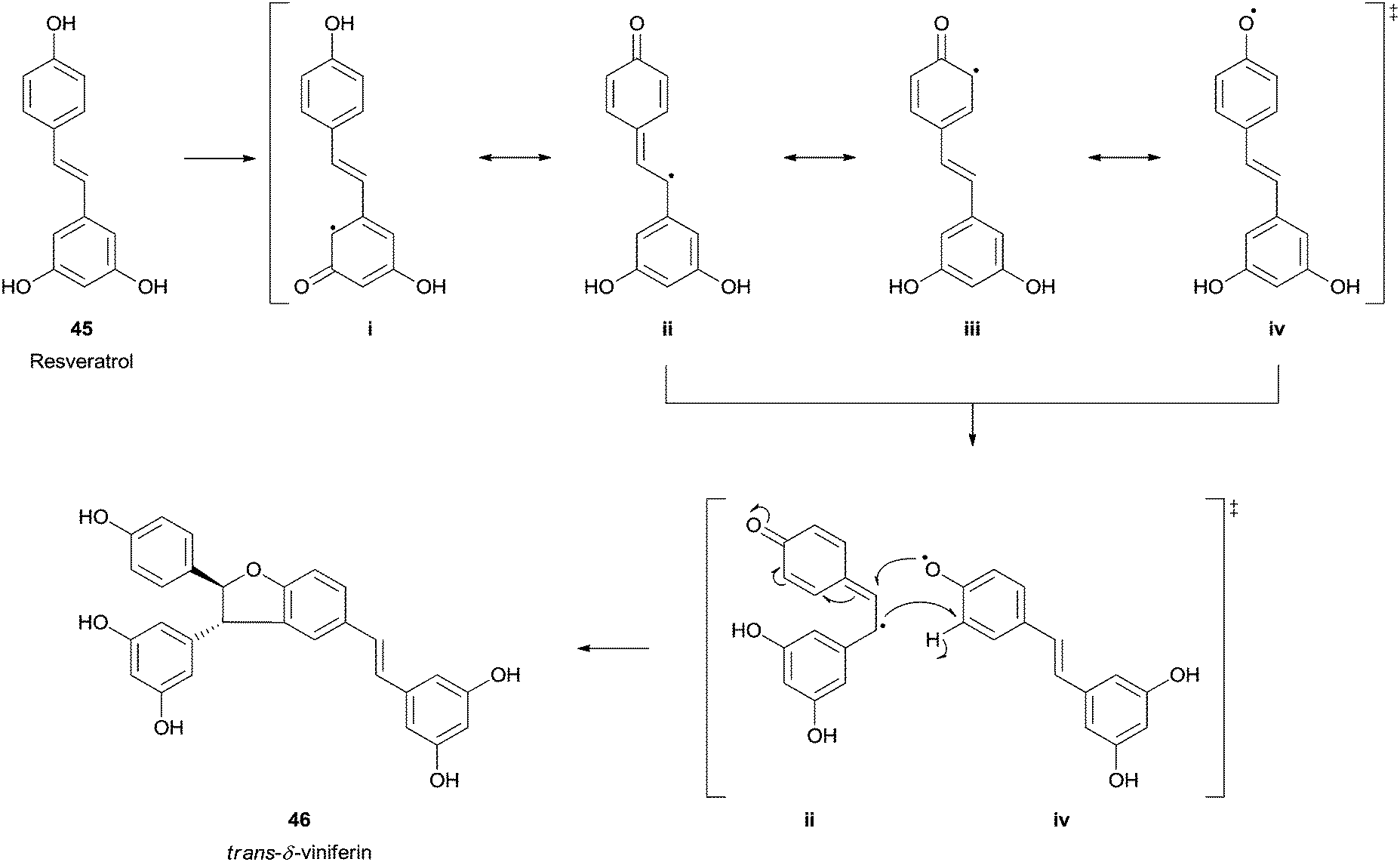 | ||
| Scheme 14 trans-δ-Viniferin 46 formation by HRP catalysed biotransformation of trans-resveratrol (45). | ||
Recently, a fascinating report described the synthesis of several resveratrol dimers from natural resveratrol using HRP.97 Interestingly, the pH adjustment allowed three different catalytic pathways. Therefore, trans-δ-viniferin (46) was obtained selectively in high yield under basic conditions, in neutral pH also leachianol F (47) and G (48), while apart from these pallidol (49) and cis-δ-viniferin (50) were produced in acidic conditions (Fig. 6). In addition, three resveratrol analogues (4-hydroxylstilbene, 3-hydroxystilbene and 3,5-dihydroxystilbene) were tested to identify the reaction mechanism and the authors found that only with the 4-hydroxylstilbene the dimerization reaction occurred. This fact supports the hypothesis that the one electron oxidation process takes place in the phenol ring of resveratrol.
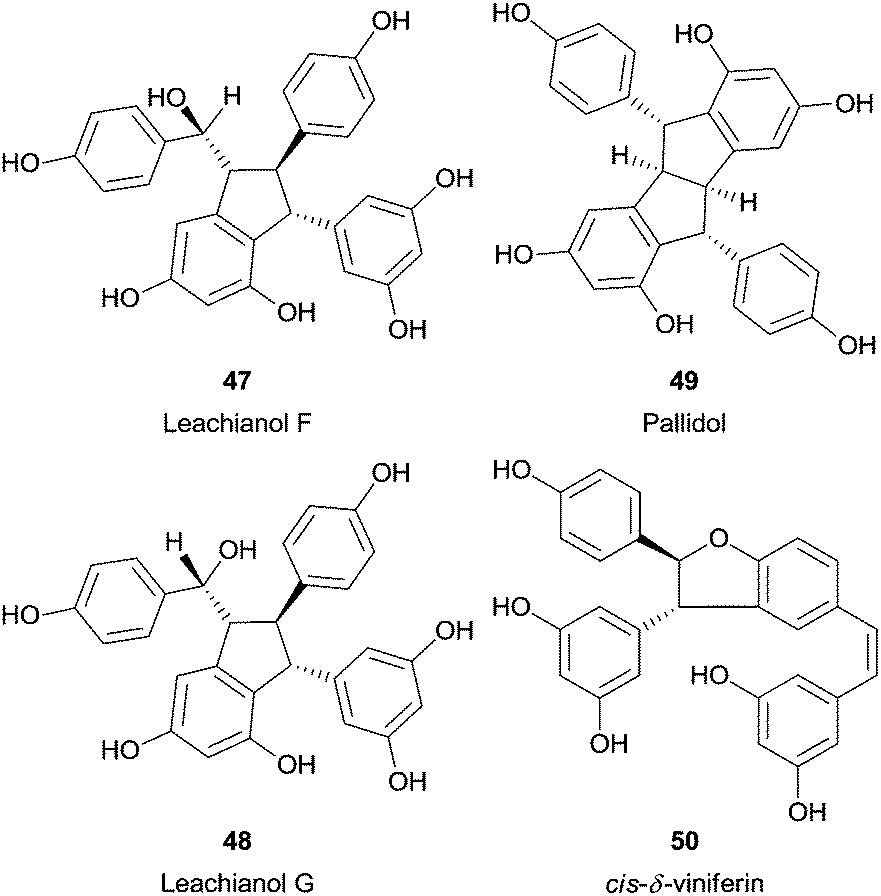 | ||
| Fig. 6 Structures of several resveratrol dimers synthesized by using HRP. | ||
An yellow pigment with important biological activity and composed mainly by hispidin (51) and its dimers were produced by medicinal fungi (Phellinus linteus and Inonotus xeranticus). Lee and Yun tried to realize the biosynthesis details of dimeric hispidins, 3,14′-bihispidinyl, hypholomine B and 1,1-distyrylpyrylethan using HRP knowing that the process should involve oxidative coupling by ligninolytic enzymes, namely laccases and peroxidases.98 In this first study on HRP-mediated dimerization of hispidin, it was only obtained the dimer 3,14′-bihispidinyl (52) (Scheme 15). The oxidative coupling at the C-3 and C-14′ positions must prevail in the process of dimerization by HRP as well as additional catalysts or substrates would be present to allow the synthesis of the other hispidin dimeric structures.
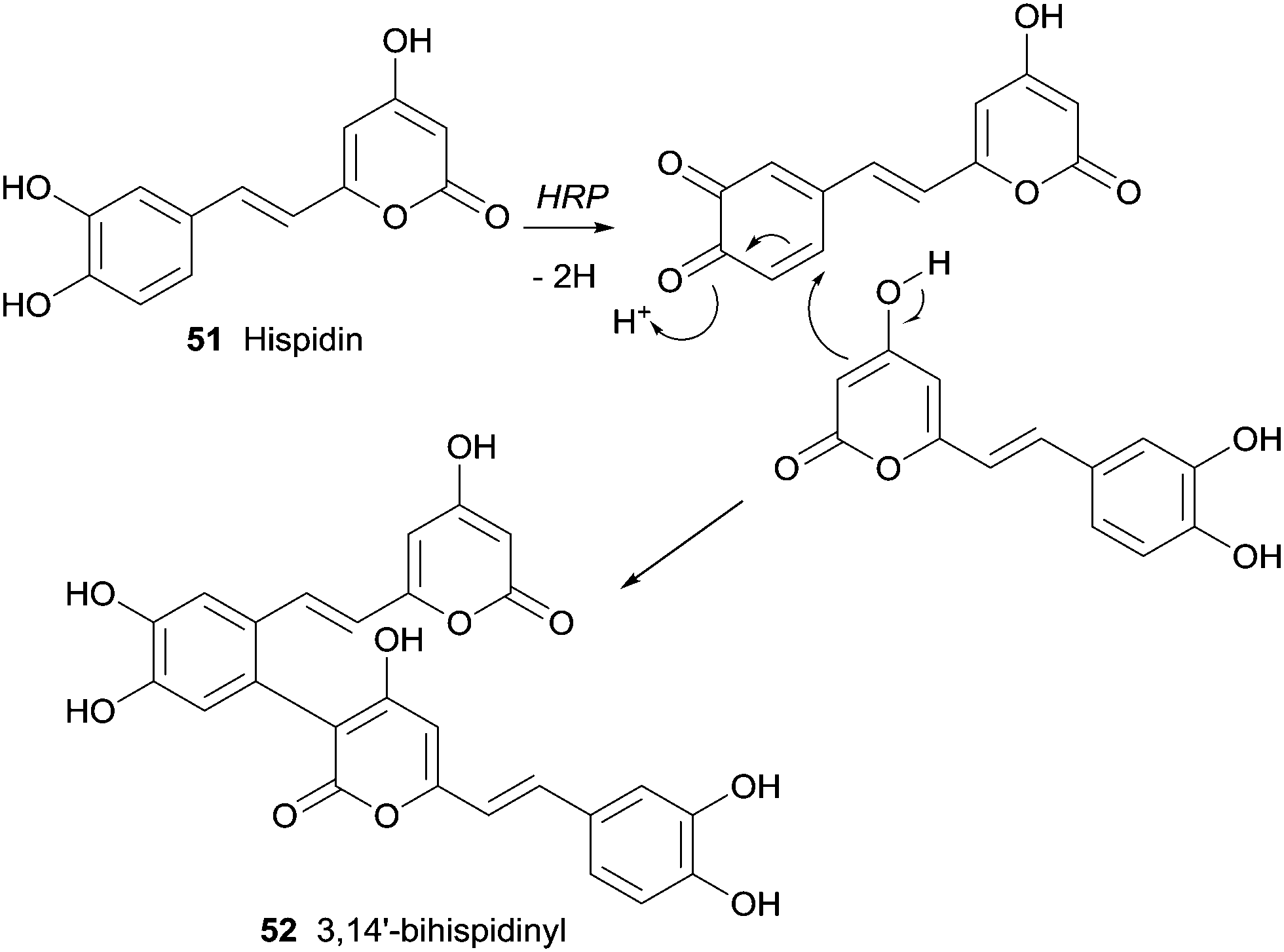 | ||
| Scheme 15 Formation of 3,14′-bihispidinyl (52) by HRP catalysed biotransformation of hispidin (51). | ||
Under optimal conditions 2-naphthol derivatives 53 can be enantioselectively oxidised to 1,1′-binaphthyl-2,2′-diol type compounds 54 by HRP and 5% H2O2 at 20 °C (Scheme 16).99 The results obtained with HRP were comparable with those obtained with the existing classical methods, as for instance, intermolecular Ullman coupling100 or nucleophilic aromatic substitution,101 in terms of yield and enantiomeric excess (ee). Contrary to the latter study, Schreier and co-workers reported the dimerization of 2-naphthol (53a) although in small yield (35%) and ee (less than 5%) (Scheme 16). Thus, they denominate HRP as an effective but unselective catalyst.102
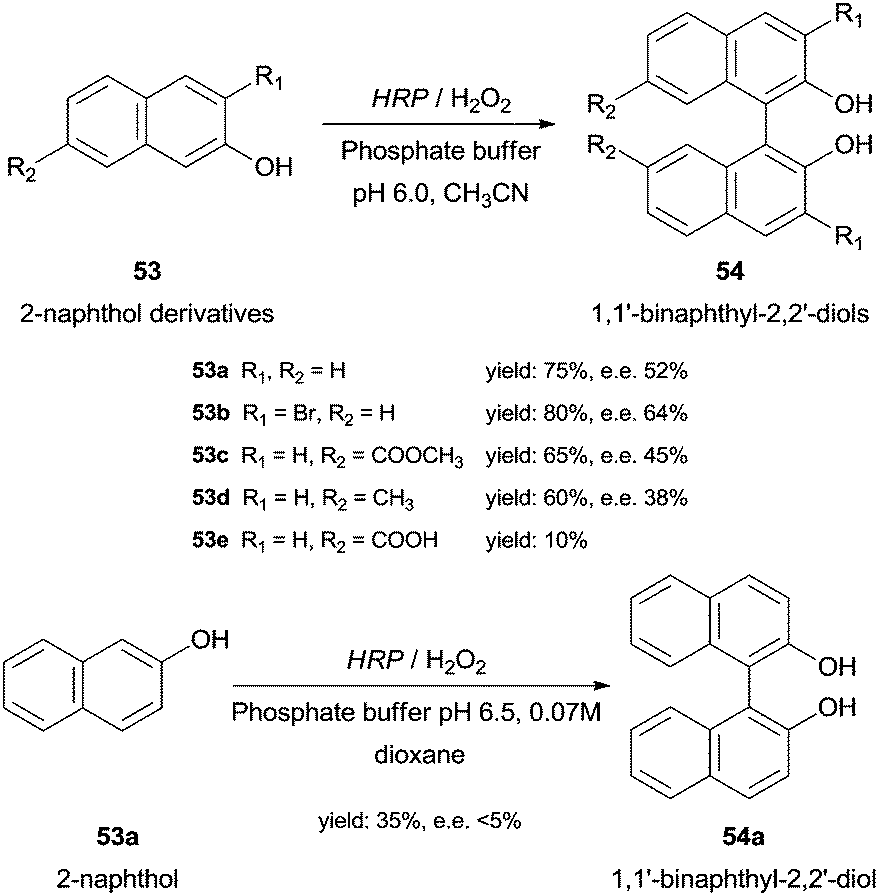 | ||
| Scheme 16 Biotransformation of 2-naphthol derivatives 53 by HRP. | ||
Another example of HRP oxidative coupling with this type of compounds involves 1-naphthol (55) and 2-hydrazono-4-thiazolines (56) to synthesize p-naphthoquinone-thiazol-2-on-azines 57 (Scheme 17).103 Taking into account the higher water solubility achieved, the thiazoline-related compounds were used in its hydrochloride form and the yields achieved were very good.
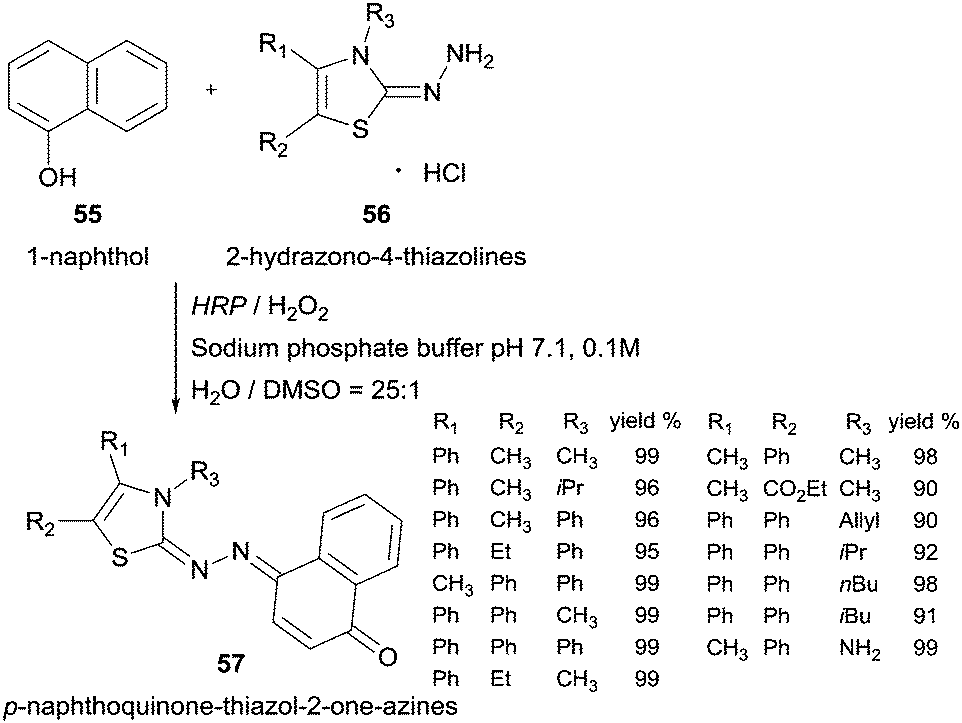 | ||
| Scheme 17 HRP catalysed coupling of 1-naphthol (55) and 2-hydrazono-4-thiazolines 56. | ||
Hydroxyanthraquinones 58 and hydroxyanthracenones 59 were tested as HRP substrates and 3,3′-bializarin (60a) and the new compound 3,3′-bipurpurin (60b) were obtained respectively from alizarin and purpurin anthraquinones (Scheme 18).104 Although quinizarin (58d) seems to be a poor substrate for HRP (10% oxidative coupling), 2,2′-biquinizarin (60c) can be obtained in good yield (80%) when quinizarin-anthracenone (59b) is used as substrate. In fact, it seems that both anthracenones and anthraquinones could be used as intermediates to synthesize bisanthraquinones and taking into account the results achieved peroxidase may play a role in the biogenesis of bisanthraquinones.
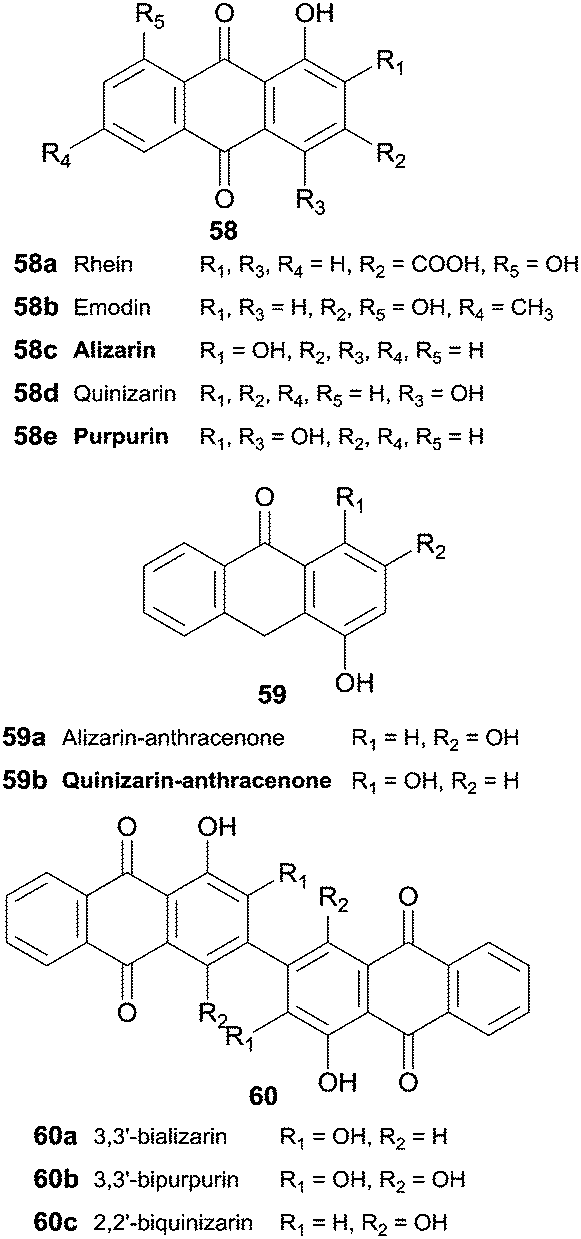 | ||
| Scheme 18 Substrates and products involved in hydroxyanthraquinones 58 and hydroxyanthracenones 59 biotransformation by HRP. | ||
5. Enzymatic hydroxylation
Several important pharmaceutical agents are hydroxylated aromatic compounds, and their syntheses usually require tedious procedures, involving protecting and deprotecting steps or catalytic systems involving toxic metals frequently accompanied by the generation of large quantities of hazardous substances. To drive toward environmentally benign synthesis, the hydroxylation of aromatics by using HRP as catalyst seems to be an attractive alternative. The first studies belong to Mason and Buhler who used this enzyme to hydroxylate aromatic compounds by molecular oxygen in the presence of dihydroxyfumaric acid (DHF) as cofactor.105 Years later Halliwell and Ahluwalia investigated the conversion of p-coumaric acid into caffeic acid in the presence of dihydroxyfumarate, stating that the latter cannot be replaced by ascorbate, H2O2, NADH, cysteine or sulfite.106 Furthermore, Mn2+ or Cu2+ completely inhibited the reaction even in low concentrations, probably due to the ability of these transition metal ions to react with the superoxide radical. The authors also concluded that this radical is generated during the oxidation of dihydroxyfumarate by HRP, reacting with H2O2 to produce hydroxyl radicals that convert p-coumaric acid to caffeic acid.Nearly thirty years ago Klibanov, Dordick and co-workers demonstrated the potential of HRP to hydroxylate aromatic compounds. From their work can be underlined the synthesis of hydroquinone and catechol from phenol,107 L-3,4-dihydroxyphenylalanine (L-DOPA) from L-tyrosine, D-(−)-3,4-dihydroxyphenylglycine from D-(−)-p-hydroxyphenylglycine and L-epinephrine (adrenaline) from L-(−)-phenylephrine.108
In the 1990s several other hydroxylation studies of phenol using HRP were reported. In fact, in the presence of DHF and oxygen, besides catechol and hydroquinone also hydroxyquinol was obtained.109 This reaction was exhaustively studied regarding namely thermodynamic and kinetic analysis.110 In fact, an important conclusion related with the catalytic function of HRP was reported. The results indicated that the production of OH˙ radicals that further react with phenol was independent of the pure catalytic cycle of the enzyme. Thus HRP must provide a source of DHF˙ radicals required for the reaction but it has no influence in the last step, the phenol hydroxylation. The thermodynamic and experimental data showed that HRP cannot be used to catalyse hydroxylation reactions instead of cytochrome P450 as suggested by some authors.
In this century, more complicated substrates were used in HRP reactions, such as violacein (61) (Fig. 7), the main pigment produced by Chromobacterium violaceum.111 The reaction result was very interesting since two simultaneous transformations occurred. In fact, it was observed the hydroxylation at the indole group and an unexpected ring contraction in the pyrrole group, giving compound 62 (Fig. 7). Furthermore, the formed compound seems to be less cytotoxic than violacein (61) which may unfortunately indicate a decrease in its antitumor activity.
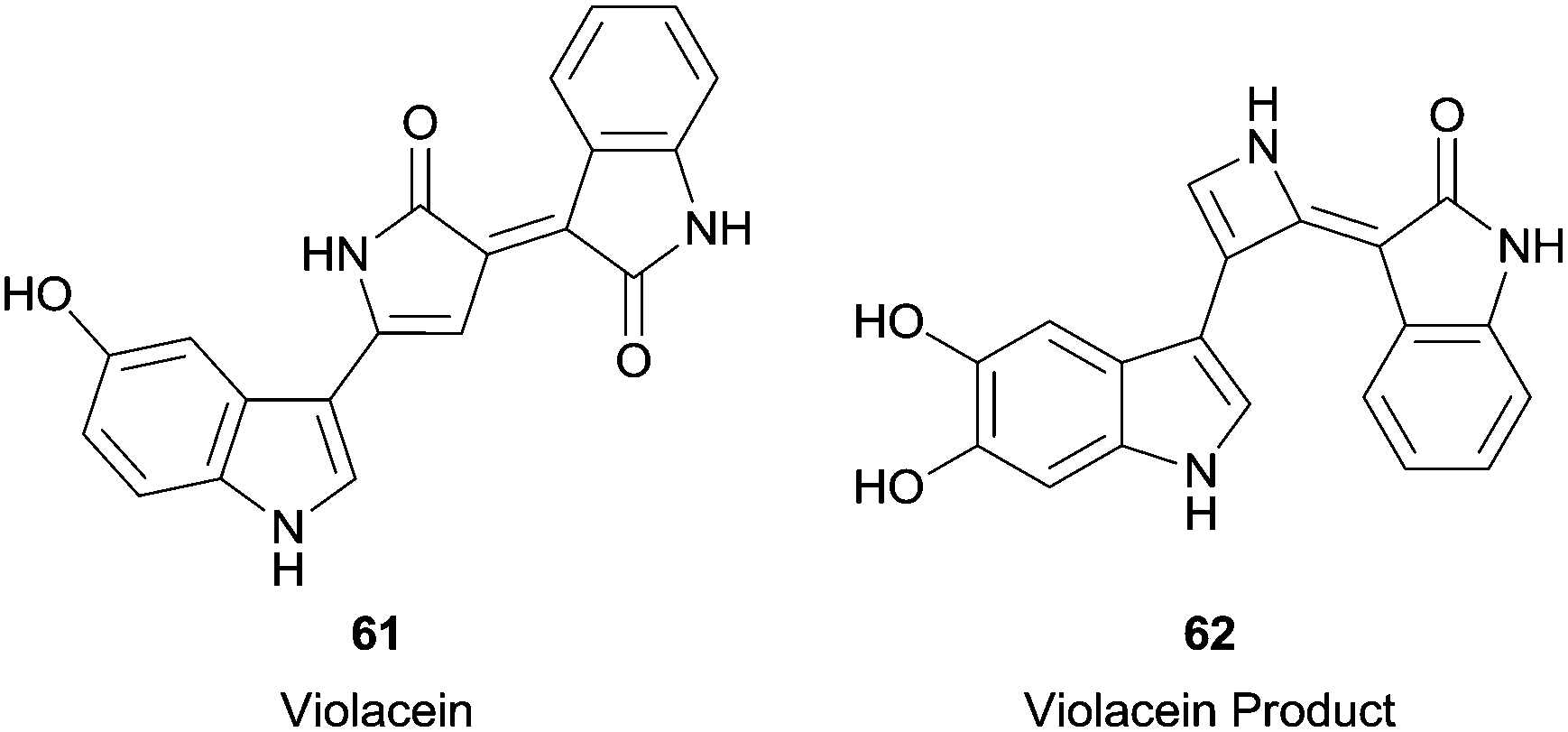 | ||
| Fig. 7 Violacein 61 and the product formed by its biotransformation with HRP. | ||
Recently, Krishna and co-workers developed a spectrophotometric assay for the quantification of peroxidase based on the hydroxylation of 4-amino-5-hydroxynapthlene-2,7-disulfonic acid monosodium salt (AHNDSA) (63) by HRP/H2O2 and posterior formation of quinone (Scheme 19).112 Despite this work represents just an example of HRP hydroxylation it allowed the development of a rapid and convenient method for peroxidase activity determination in plant extracts with comparable values of catalytic efficiencies of the well-known guaiacol method. Useful in this case is the stable chromogenic product obtained with intense yellow colour and high molar extinction coefficient.
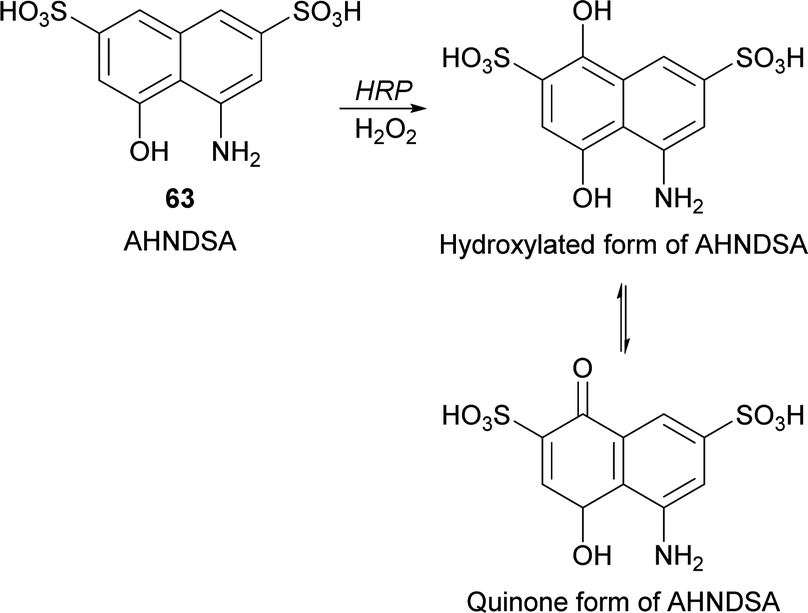 | ||
| Scheme 19 Hydroxylation of 4-amino-5-hydroxynapthlene-2,7-disulfonic acid monosodium salt (AHNDSA) (63) by HRP/H2O2. | ||
Although the examples presented herein show that HRP may be used to some extent in hydroxylation reactions, this enzyme clearly was not the preferential to catalyse this kind of reaction even in the group of peroxidases.113 Regarding this fact, the catalytic cycle and conformational structure themselves seem to be the principal barriers to HRP action. In fact, this enzyme undoubtedly prefers the sequential one-electron transfer processes instead of two-electron transfer reactions where ferryl oxygen is transferred to the substrates.3,114 Furthermore, in conformational terms there is a hindrance to direct oxygen transfer since the access to the reactive oxoferryl species is obstructed by bulky amino acids in the distal pocket of the enzyme (e.g. His42) being the active site sterically restricted to substrates.3,113–115 To overcome these problems some groups have tried to apply genetic engineering methodologies, namely site-directed mutagenesis, in order to improve the ability to transfer oxygen. The application of this technique allows the substitution of some amino acid residues and the achievement of several mutants. In this context, some of them should ideally have cytochrome P450 like properties. Nonetheless their hydroxylation capability is limited.5a,b,116
6. Enzymatic nitration and sulfoxidation
The synthesis of nitro compounds is useful taking into consideration that a nitro group can be an excellent scaffold to obtain other derivatives. The classic nitration of phenols usually involves the use of nitric acid diluted in water or acetic acid, which are harmful to the environment. Thereby, as well as other hemoproteins, HRP has been used as a green catalyst that is able to oxidize NO2− in the presence of H2O2 to nitrate aromatic compounds. Dai and co-workers studied a biocatalytic nitration of phenol (64a) and m-cresol (64b) in the presence of hydrogen peroxide and sodium nitrite under mild conditions (room temperature at pH 7.0).117 In the case of phenol (64a), 4-nitrophenol (65a) (14%) and 2-nitrophenol (66a) (12%) were the main products obtained. Some minor by-products like hydroquinone and catechol were also obtained. In respect to m-cresol (64b), the products obtained were 4-nitro-3-methylphenol (65b) (19%) and 2-nitro-5-methylphenol (66b) (30%), showing that nitration by HRP occurs at the same positions in phenol type structures (Scheme 20).117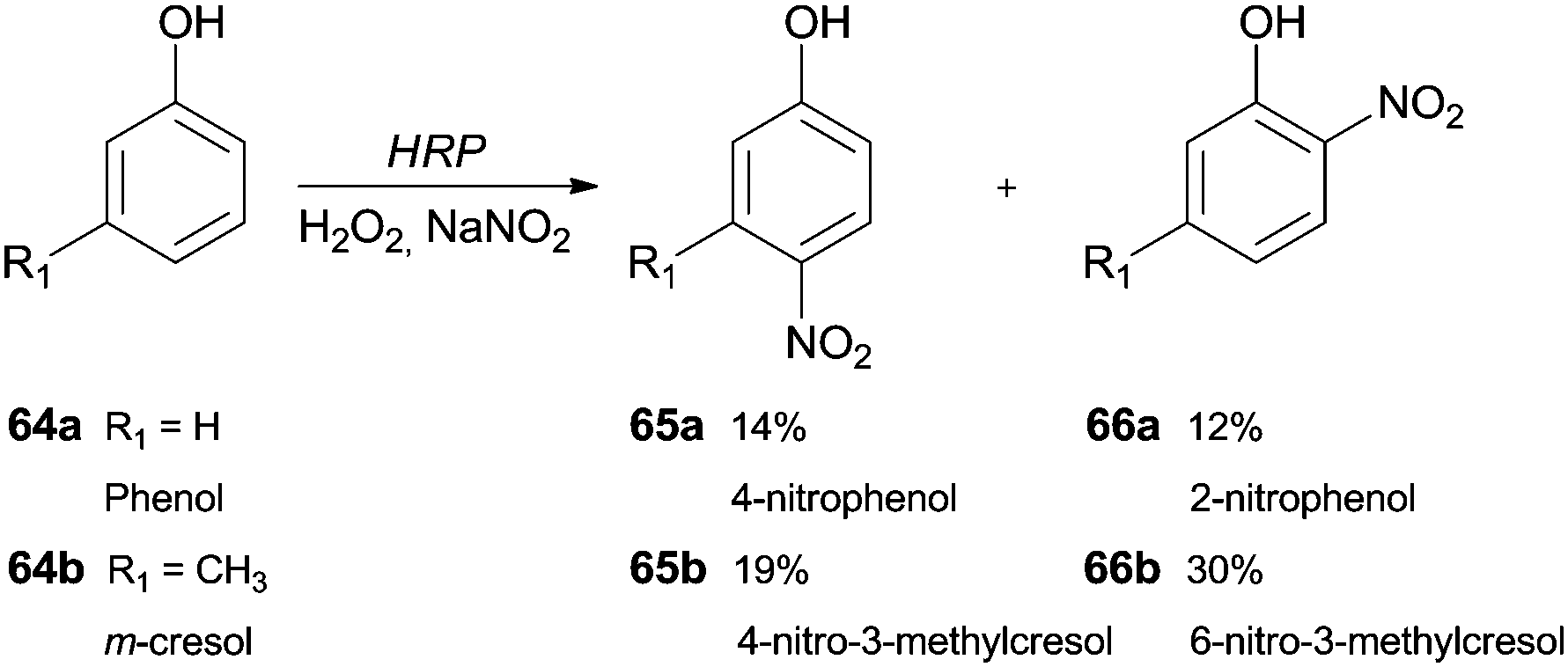 | ||
| Scheme 20 Nitration reactions performed by HRP/H2O2/NaNO2 system with phenol (64a) and m-cresol (64b) as substrates. | ||
Similar approaches have been applied for the nitration of 4′-hydroxy-3′-methylacetophenone (67)118 and 4-hydroxycinnamic acid.119 The authors reported that nitration was directed to the ortho-position of the hydroxyl group, being obtained 4′-hydroxy-3′-methyl-5′-nitroacetophenone (68) and 4-hydroxy-3-nitrocinnamic acid, respectively. However, in the case of 4′-hydroxy-3′-methylacetophenone (67) it was also obtained the 2-methyl-4-nitrophenol (69) (Scheme 21); this para-nitration only occurred because a simultaneously elimination of the acetyl group took place.118
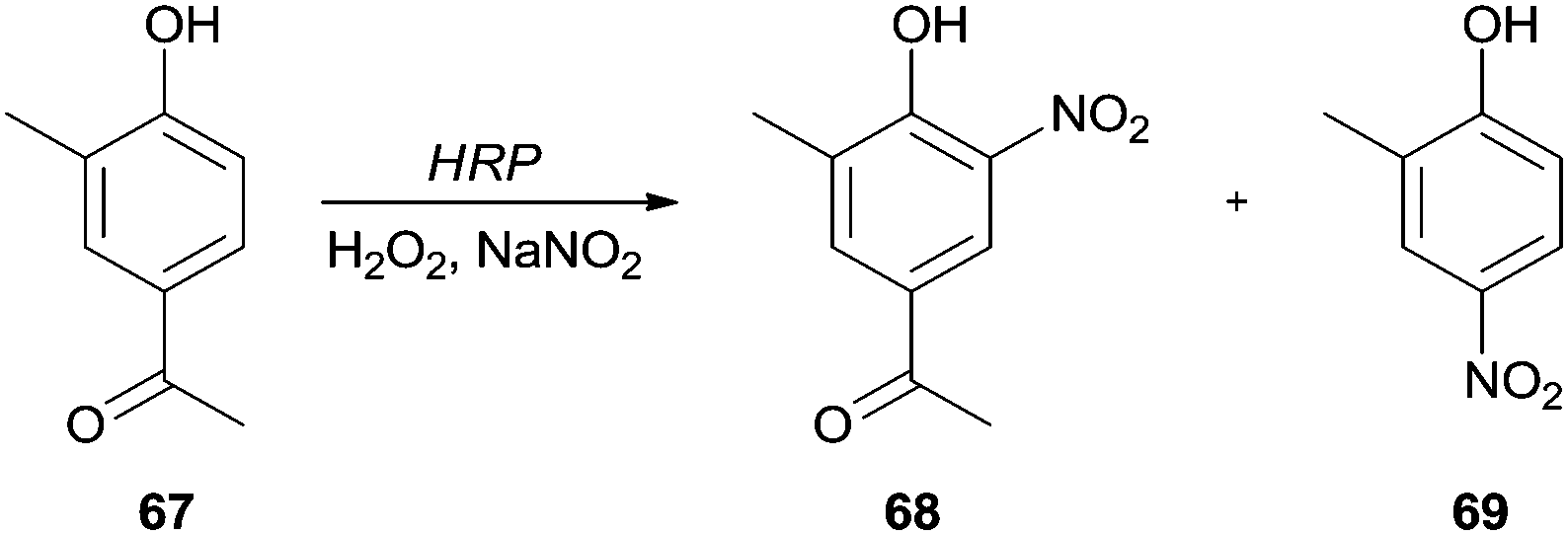 | ||
| Scheme 21 Nitration of 4′-hydroxy-3′-methylacetophenone (67) by the HRP/H2O2/NaNO2 system. | ||
Besides the use of simple phenol derivatives more complex substrates were also used, from which the use of natural compounds can be highlighted. For instance, the enzymatic nitration of tryptophan derivatives 70 (Fig. 8) by the system HRP/NO2−/H2O2 proved to be dependent on the nitrite concentration and also selective to positions 4-, 6-, 7- and N- of the indole ring.120 In fact, the study of the nitration of amino acid derivatives is quite important. HRP was also used to oxidize nitrite in the presence of hydrogen peroxide to allow the nitration of both free tyrosine and tyrosine residues in proteins, the most typical target of nitrating species.121
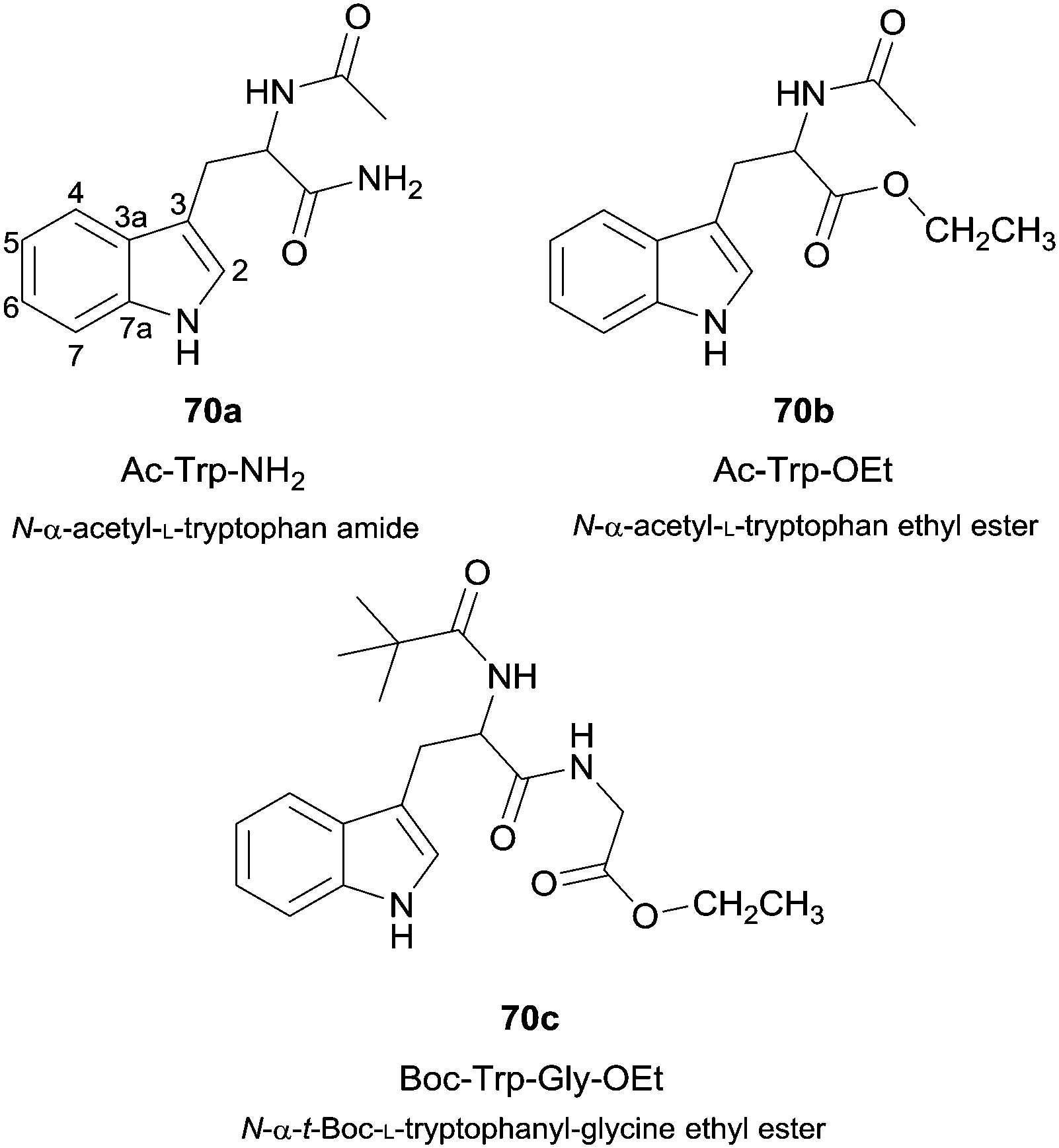 | ||
| Fig. 8 Tryptophan derivatives used as substrates in HRP catalysed nitration reactions. | ||
Another interesting example involves the nitration of one of the principal naturally occurring estrogens, the 17β-estradiol. Since geno- and cytotoxic mechanisms in oxidative stress-based diseases may be due to nitration reactions targeted to cellular constituents, studying a HRP/H2O2/NO2− system to 17β-estradiol nitration could give chemical information about the mechanism of impairment of estrogen functions under oxidative/nitrosative conditions. Pezzella and co-workers studied the 17β-estradiol nitration with such system and obtained 2-nitroestradiol (71), 4-nitroestradiol (72) and 2,4-dinitroestradiol (73) when an excess of nitrite was used (Fig. 9).122 The nitration seems to be regioselective for C-4 position under slow generation of nitrating species and that estradiol is at least as susceptible to nitration as tyrosine, the nitratable biological target par excellence.
 | ||
| Fig. 9 Structures obtained in the 17β-estradiol nitration by the HRP/H2O2/NO2− system. | ||
The same group also studied the pathways of dopamine (74) oxidation using HRP as catalyst among other reagent systems tested.123 They firstly found that the neurotoxin 6-hydroxydopamine (75) was formed by HRP/H2O2 system.123b However, the presence of nitrite ions (NO2−) beyond H2O2 led also to the formation of 6-nitrodopamine (76) due to the reaction of dopamine (74) with NO-derived species (Fig. 10).123a Additional control experiments revealed that any dopamine product of nitration reaction was detectable with various NO2− concentrations when neither HRP nor H2O2 were present. The authors also gave special attention to pH in order to avoid that it dropped below 7.4 and consequent nitration of dopamine occurs by the presence of HNO2 in acidic medium. Taking into account the two possible products formed it was found that with fixed amounts of H2O2 the formation of the nitro derivative increase when concentration of NO2− increase whereas the formation of 6-hydroxydopamine (75) decreases. The system D-glucose/glucose oxidase was used to promote a low and constant flux of H2O2 in situ generated. Other experiences suggested that reactive nitrogen radicals are not involved in 6-nitrodopamine (76) formation being the nucleophilic attack of NO2− to dopamine quinone the main reason for the reaction, whereas diverse H2O2-dependent pathways seems to be involved in 6-hydroxydopamine (75) formation. Biological experiences were done to get some useful information for the study of oxidative stress- and nitric oxide-induced neurotoxicity related to neurodegenerative disorders.
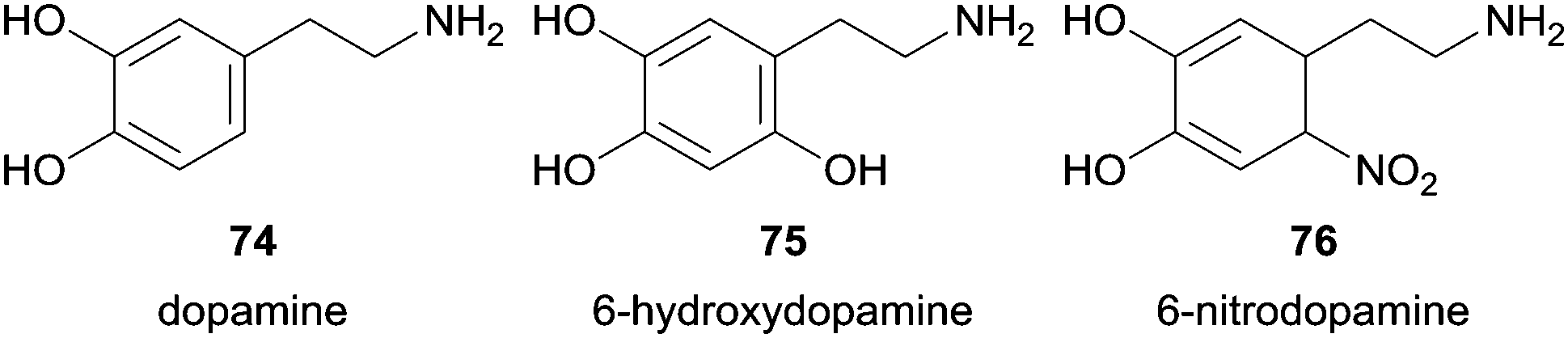 | ||
| Fig. 10 Products 75, 76 obtained in nitration reaction of dopamine (74). | ||
Additionally we found in the literature other class of compounds used as substrate in a nitration reaction in the presence of HRP. Although they are not phenolic compounds, the obtained result is an unexpected chain rearrangement of linoleic acid metabolite, when exposed to nitrating agents that can elucidate physiological important pathways of lipid modification. At mild conditions (pH 7.4 and room temperature) and in the presence of HRP/H2O2/NO2− system, the (13S,9Z,11E)-13-hydroxyoctadeca-9,11-dienoic acid (77) was transformed in two diastereomeric products, methyl (12S)-10,11-epoxy-12-hydroxy-9-nitromethylheptadecanoates 78, 79 (Scheme 22).124
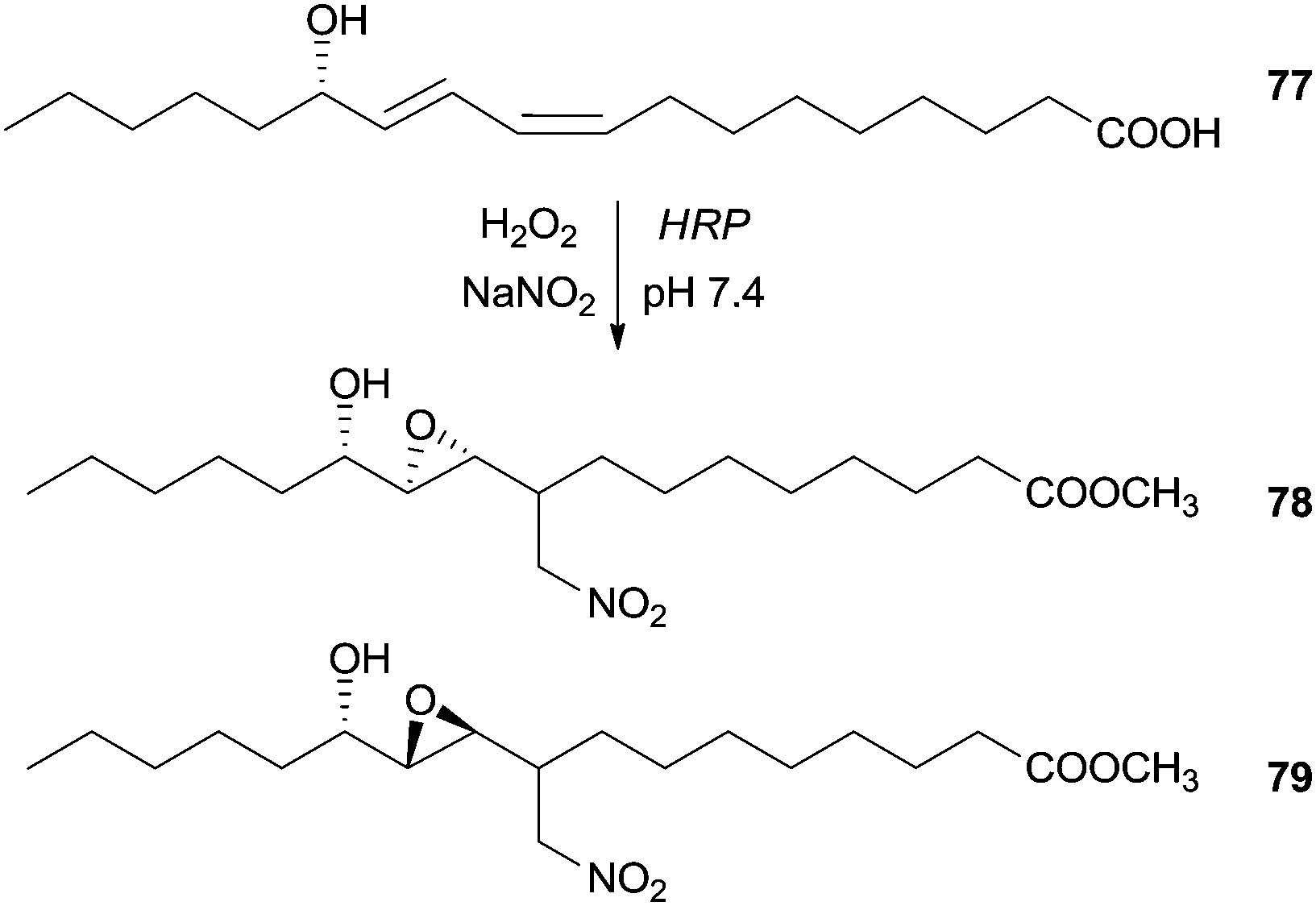 | ||
| Scheme 22 Nitration reaction of (13S,9Z,11E)-13-hydroxyoctadeca-9,11-dienoic acid (77) by the HRP/H2O2/NO2− system. | ||
Besides the above mentioned nitration reactions, HRP can catalyse several asymmetric processes leading to the formation of valuable products as sulfoxides being many of them chiral. Kumar and co-workers claimed that theoretically HRP should be as reactive as cytochrome P450 in sulfoxidation reactions, although HRP acts mainly as an electron sink and not as an oxygen-transfer. Compared with P450 enzymes, HRP reactivity is normally slow, due predominantly to conformational restrictions namely the smaller substrate-binding pocket in HRP whereby this result proved quite interesting.125 Although there are several studies concerning asymmetric sulfoxidation by HRP there is no agreement on the best experimental method regarding the yield and the ee of the formed sulfoxide. Some conditions as concentrations of the sulfide and enzyme, pH or the way how H2O2 is added to the reaction may significantly change the results. Since the early 1990s a series of papers concerning sulfoxidations developed mainly by the research groups of Colonna and Ozaki highlight that HRP showed some lack of selectivity (Scheme 23).5c,126,127 A simple modification on the HRP structure, as the replacement of phenylalanine in the native structure by a leucine, seems to enhance the selectivity towards sulfoxidation reactions.5c,127
 | ||
| Scheme 23 Substrates used in sulfoxidations reactions with HRP as catalyst. | ||
Tuynman and co-workers studied the asymmetric sulfoxidation of thioanisole with HRP as catalyst and found that the process depend strongly on the experimental conditions. In fact, thioanisole could be completely converted to the corresponding sulfoxide (100% yield and 60% ee) (S)-sulfoxide using a slow continuous influx of H2O2 for 48 hours. This approach for H2O2 addition allowed the achievement of high ee values but was also necessary to prevent the accumulation of H2O2 and the formation of catalytically inactive species.128 In order to avoid that HRP becomes readily inactive under harsh conditions as organic solvent and high oxidant and sulfide concentrations, Ferrer and co-workers studied the encapsulation of HRP in a sol–gel matrix to obtain a heterogeneous catalyst.129 They used the enantioselective sulfoxidation of thioanisole as the model reaction to investigate the oxygen-transfer catalytic property of this system, being acetonitrile chosen among other solvents since it quickly causes deterioration on enzyme activity allowing a good assay for enzyme protection. Although free HRP showed the ability to perform the reaction, the total turnover number (i.e. mmol product/mmol catalyst) increased up to six-fold with the encapsulated system. Furthermore, the sulfoxide selectivity and the ee also increased greatly upon encapsulation, being possible to recycle the heterogeneous catalyst by simple filtration in successive runs. Also Xie and co-workers reported an increased stereoselectivity for the model sulfide substrate thioanisole when the enzyme is co-lyophilized in the presence of lypoprotectants or ligands.130
HRP was also used to catalyse the sulfoxidation of promethazine, being the reaction studied as a function of pH as well as promethazine and H2O2 concentration.131 The authors found that oxygen is required, superoxide dismutase has no effect on the sulfoxidation of promethazine, but ascorbic acid and glutathione inhibited the reaction. The sulfoxide derivatives that are part of some of the main metabolites of a few drugs, usually lose their activity after sulfoxidation. Interestingly, it seems that this reaction does not affect significantly the neuroleptic effect of the original compound suggesting that it could takes place in vivo forming the same active species for the aforementioned biological effect.
HRP has also the ability to catalyse thiosulfinations, which means that it can stereoselectively catalyse the monooxidation of organic disulfides to thiosulfinates. Dzyuba and Klibanov showed that HRP could catalyse the mentioned reaction and exhibit specificity.132 In fact, the thiosulfination of 1,2-dithiane was much faster and more stereoselective than that of di-tert-butyl disulfide when the reaction took place under buffer conditions. The lower solubility of di-tert-butyl disulfide in aqueous media could explain that result. When the process was performed in methanol the rate of products formation increases due probably to the greater solubility of the disulfide substrates in the organic media, although there is a pronounced drop in stereoselectivity.
7. Conclusions
Green chemistry principles have challenged the synthetic organic chemists to develop new synthetic routes with low waste methods and consequently improve the environmental performance of many syntheses. Among these are the development of biocatalytic processes from where the use of HRP is certainly one of the most fruitfully examples. As illustrated herein HRP greatly increase flexibility in synthetic design. It is an alternative for the efficient synthesis of several types of polymers as well as in the synthesis of pharmaceutical agents. For instance the stereo- and regioselective introduction of functional groups such as hydroxyl is tremendously important in the synthesis of drug molecules. Further uses in academic labs and future large scale applications will expand its use and consequently new green chemical synthesis will be developed.Acknowledgements
Thanks are due to the University of Aveiro, Fundação para a Ciência e a Tecnologia (FCT, Portugal) and the European Union, QREN, FEDER, COMPETE, for funding the Organic Chemistry Research Unit (QOPNA) (project PEst-C/QUI/UI0062/2013; FCOMP-01-0124-FEDER-037296).Notes and references
- (a) K. Faber, Biotransformations in Organic Chemistry, Springer-Verlag, Berlin, 5th edn, 2004 Search PubMed; (b) J. Tao and R. Kazlauskas, Biocatalysis for Green Chemistry and Chemical Process Development, John Wiley & Sons, New Jersey, 2011 Search PubMed; (c) K. Drauz and H. Waldmann, Enzyme Catalysis in Organic Synthesis-A Comprehensive Handbook, Wiley-VCH, Weinheim, 2nd edn, 2002 Search PubMed.
- M. Hamid and R. Khalilur, Food Chem., 2009, 115, 1177–1186 CrossRef CAS PubMed.
- N. C. Veitch, Phytochemistry, 2004, 65, 249–259 CrossRef CAS PubMed.
- (a) G. Meunier and B. Meunier, J. Am. Chem. Soc., 1985, 107, 2558–2560 CrossRef CAS; (b) M. Skarpeli-Liati, S. G. Pati, J. Bolotin, S. N. Eustis and T. B. Hofstetter, Environ. Sci. Technol., 2012, 46, 7189–7198 CrossRef CAS PubMed; (c) G. L. Kedderis and P. F. Hollenberg, J. Biol. Chem., 1984, 259, 3663–3668 CAS; (d) N. W. Gaikwad and W. J. Bodell, Free Radical Biol. Med., 2012, 52, 340–347 CrossRef CAS PubMed.
- (a) M. I. Savenkova, S. L. Newmyer and P. R. O. de Montellano, J. Biol. Chem., 1996, 271, 24598–24603 CrossRef CAS PubMed; (b) M. Tanaka, K. Ishimori, M. Mukai, T. Kitagawa and I. Morishima, Biochemistry, 1997, 36, 9889–9898 CrossRef CAS PubMed; (c) S.-I. Ozaki and P. R. O. de Montellano, J. Am. Chem. Soc., 1995, 117, 7056–7064 CrossRef CAS.
- (a) S. Preethi, A. Anumary, M. Ashokkumar and P. Thanikaivelan, SpringerPlus, 2013, 2, 1–8 CrossRef PubMed; (b) V. A. Cooper and J. A. Nicell, Water Res., 1996, 30, 954–964 CrossRef CAS; (c) Q. Husain, Rev. Environ. Sci. Biotechnol., 2010, 9, 117–140 CrossRef CAS.
- (a) T. Ruzgas, E. Csöregi, J. Emnéus, L. Gorton and G. Marko-Varga, Anal. Chim. Acta, 1996, 330, 123–138 CrossRef CAS; (b) A. K. M. Kafi, G. Wu and A. Chen, Biosens. Bioelectron., 2008, 24, 566–571 CrossRef CAS PubMed; (c) M. A. Alonso-Lomillo, O. Domínguez-Renedo, L.-D. T.-D. Román and M. J. Arcos-Martínez, Anal. Chim. Acta, 2011, 688, 49–53 CrossRef CAS PubMed; (d) J. Jia, B. Wang, A. Wu, G. Cheng, Z. Li and S. Dong, Anal. Chem., 2002, 74, 2217–2223 CrossRef CAS; (e) Y. Hao, B. Zhou, F. Wang, J. Li, L. Deng and Y.-N. Liu, Biosens. Bioelectron., 2014, 52, 422–426 CrossRef CAS PubMed; (f) C. Zhang, T. Wei, M. Han, W. Tu and Z. Dai, Electrochem. Commun., 2012, 22, 133–136 CrossRef CAS PubMed.
- M. F. Machado and J. Saraiva, J. Mol. Catal. B: Enzym., 2002, 19–20, 451–457 CrossRef CAS.
- L. A. Planche, Bull. de Pharmacie, 1810, 2, 578–580 Search PubMed.
- (a) B. Y. Yang, J. S. S. Gray and R. Montgomery, Carbohydr. Res., 1996, 287, 203–212 CrossRef CAS; (b) K. G. Welinder, FEBS Lett., 1976, 72, 19–23 CrossRef CAS.
- (a) H. B. Dunford, in Peroxidases in Chemistry and Biology, ed. J. Everse, K. E. Everse and M. B. Grisham, CRC Press, Boca Raton, FL, 1991, vol. 2, pp. 1–24 Search PubMed; (b) B. Meunier, in Comprehensive Coordination Chemistry II, ed. E. J. McClaverty and T. J. Meyer, Wiley–VCH, New York, 2003, pp. 261–280 Search PubMed; (c) H. B. Dunford, Heme peroxidases, Wiley–VCH, New York, 1999 Search PubMed; (d) P. R. O. Montellano, Annu. Rev. Pharmacol., 1992, 32, 89–107 CrossRef PubMed; (e) P. O. de Montellano, in Biocatalysis Based on Heme Peroxidases, ed. E. Torres and M. Ayala, Springer, Berlin Heidelberg, 2010, pp. 79–107 Search PubMed; (f) J. N. Rodríguez-López, D. J. Lowe, J. Hernández-Ruiz, A. N. P. Hiner, F. García-Cánovas and R. N. F. Thorneley, J. Am. Chem. Soc., 2001, 123, 11838–11847 CrossRef PubMed.
- H. B. Dunford and J. S. Stillman, Coord. Chem. Rev., 1976, 19, 187–251 CrossRef CAS.
- (a) R. Santucci, E. Laurenti, F. Sinibaldi and R. P. Ferrari, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2002, 1596, 225–233 CrossRef CAS; (b) L. Haifeng, L. Yuwen, C. Xiaomin, W. Zhiyong and W. Cunxin, J. Therm. Anal. Calorim., 2008, 93, 569–574 Search PubMed; (c) S. Asad, S.-F. Torabi, M. Fathi-Roudsari, N. Ghaemi and K. Khajeh, Int. J. Biol. Macromol., 2011, 48, 566–570 CrossRef CAS PubMed; (d) K. Chattopadhyay and S. Mazumdar, Biochemistry, 1999, 39, 263–270 CrossRef PubMed.
- A. M. Azevedo, D. M. F. Prazeres, J. M. S. Cabral and L. S. P. Fonseca, J. Mol. Catal. B: Enzym., 2001, 15, 147–153 CrossRef CAS.
- (a) L. E. S. Brink, J. Tramper, K. C. A. M. Luyben and K. Van 't Riet, Enzyme Microb. Technol., 1988, 10, 736–743 CrossRef CAS; (b) J. S. Dordick, Enzyme Microb. Technol., 1989, 11, 194–211 CrossRef CAS.
- J.-Z. Liu, T.-L. Wang, M.-T. Huang, H.-Y. Song, L.-P. Weng and L.-N. Ji, Protein Eng., Des. Sel., 2006, 19, 169–173 CrossRef CAS PubMed.
- S. M. Siegel and K. Roberts, Origins Life Evol. Biospheres, 1968, 1, 131–134 CrossRef CAS.
- M.-R. Kula, in Enzyme Catalysis in Organic Synthesis, ed. H. W. Karlheinz Drauz, Wiley-VCH, Mörlenbach, 2002, pp. 1–39 Search PubMed.
- G. Grogan, Practical Biotransformations: A Beginner's Guide, Wiley, Cornwall, 2009 Search PubMed.
- Z. Temoçin and M. Yiğitoğlu, Bioprocess Biosyst. Eng., 2009, 32, 467–474 CrossRef PubMed.
- H.-Y. Song, J.-H. Yao, J.-Z. Liu, S.-J. Zhou, Y.-H. Xiong and L.-N. Ji, Enzyme Microb. Technol., 2005, 36, 605–611 CrossRef CAS PubMed.
- (a) K. Hernandez, A. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, Curr. Org. Chem., 2012, 16, 2652–2672 CrossRef CAS; (b) M. Puiu, M. Constantinovici, I. Babaligea, A. Raducan, C. Olmazu and D. Oancea, Chem. Eng. Technol., 2010, 33, 414–420 CrossRef CAS PubMed; (c) B. Valderrama, M. Ayala and R. Vazquez-Duhalt, Chem. Biol., 2002, 9, 555–565 CrossRef CAS; (d) J. Hernandez-Ruiz, M. B. Arnao, A. N. P. Hiner, F. Garcia-Canovas and M. Acosta, Biochem. J., 2001, 354, 107–114 CrossRef CAS; (e) M. B. Arnao, M. Acosta, J. A. del Rio and F. García-Cánovas, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1990, 1038, 85–89 CrossRef CAS; (f) S. A. Adediran and A.-M. Lambeir, Eur. J. Biochem., 1989, 186, 571–576 CrossRef CAS PubMed.
- (a) A. N. P. Hiner, J. Hernández-Ruiz, G. Acute, F. Cánovas, A. T. Smith, M. B. Arnao and M. Acosta, Eur. J. Biochem., 1995, 234, 506–512 CAS; (b) B. J. Ryan and C. Ó'Fágáin, Biochimie, 2007, 89, 1029–1032 CrossRef CAS PubMed.
- (a) F. Jia, B. Narasimhan and S. Mallapragada, Biotechnol. Bioeng., 2014, 111, 209–222 CrossRef CAS PubMed; (b) A. Homaei, R. Sariri, F. Vianello and R. Stevanato, J. Chem. Biol., 2013, 6, 185–205 CrossRef PubMed; (c) R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC; (d) D. N. Tran and K. J. Balkus, ACS Catal., 2011, 1, 956–968 CrossRef CAS; (e) R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS PubMed.
- (a) O. Barbosa, R. Torres, C. Ortiz, Á. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, Biomacromolecules, 2013, 14, 2433–2462 CrossRef CAS PubMed; (b) R. C. Rodrigues, C. Ortiz, A. Berenguer-Murcia, R. Torres and R. Fernandez-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC; (c) R. Singh, M. Tiwari, R. Singh and J.-K. Lee, Int. J. Mol. Sci., 2013, 14, 1232–1277 CrossRef CAS PubMed; (d) C. Garcia-Galan, Á. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS PubMed; (e) C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS PubMed.
- (a) N. N. Rao, S. Lütz, K. Seelbach and A. Liese, in Industrial Biotransformations, ed. A. Liese, K. Seelbach and C. Wandrey, Wiley-VCH, Weinheim, 2006, pp. 115–145 Search PubMed; (b) W. A. Loughlin, Bioresour. Technol., 2000, 74, 49–62 CrossRef CAS.
- (a) Y. Jiang, W. Tang, J. Gao, L. Zhou and Y. He, Enzyme Microb. Technol., 2014, 55, 1–6 CrossRef CAS PubMed; (b) Y. Jiang, C. Cui, L. Zhou, Y. He and J. Gao, Ind. Eng. Chem. Res., 2014, 53, 7591–7597 CrossRef CAS; (c) R. Xu, C. Chi, F. Li and B. Zhang, Bioresour. Technol., 2013, 149, 111–116 CrossRef CAS PubMed; (d) S. A. Mohamed, A. L. Al-Malki, T. A. Kumosani and R. M. El-Shishtawy, Int. J. Biol. Macromol., 2013, 60, 295–300 CrossRef CAS PubMed; (e) B.-W. Park, K.-A. Ko, D.-Y. Yoon and D.-S. Kim, Enzyme Microb. Technol., 2012, 51, 81–85 CrossRef CAS PubMed; (f) Y. Li, H. Zhang and F. Cao, J. Sol-Gel Sci. Technol., 2011, 58, 156–161 CrossRef CAS.
- (a) S. Asad, B. Dabirmanesh and K. Khajeh, Biotechnol. Appl. Biochem., 2014, 61, 226–229 CrossRef CAS PubMed; (b) L. Hassani and R. Nourozi, Appl. Biochem. Biotechnol., 2014, 172, 3558–3569 CrossRef CAS PubMed; (c) S. Asad, K. Khajeh and N. Ghaemi, Appl. Biochem. Biotechnol., 2011, 164, 454–463 CrossRef CAS PubMed; (d) B. Ryan and C. O'Fagain, BMC Biotechnol., 2007, 7, 86 CrossRef PubMed.
- (a) N. J. Turner, Nat. Chem. Biol., 2009, 5, 567–573 CrossRef CAS PubMed; (b) P. A. Dalby, Curr. Opin. Struct. Biol., 2003, 13, 500–505 CrossRef CAS; (c) N. J. Turner, Trends Biotechnol., 2003, 21, 474–478 CrossRef CAS PubMed; (d) U. T. Bornscheuer and M. Pohl, Curr. Opin. Chem. Biol., 2001, 5, 137–143 CrossRef CAS.
- (a) J. Rocha-Martin, S. Velasco-Lozano, J. M. Guisan and F. Lopez-Gallego, Green Chem., 2014, 16, 303–311 RSC; (b) J. Sun, J. Ge, W. Liu, M. Lan, H. Zhang, P. Wang, Y. Wang and Z. Niu, Nanoscale, 2014, 6, 255–262 RSC.
- (a) S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353 CrossRef CAS PubMed; (b) F. Hollmann and I. W. C. E. Arends, Polymers, 2012, 4, 759–793 CrossRef PubMed.
- L. Zhang, W. Zhao, Z. Ma, G. Nie and Y. Cui, Eur. Polym. J., 2012, 48, 580–585 CrossRef CAS PubMed.
- L. Zhang, W. Zhao, H. Chen and Y. Cui, J. Mol. Catal. B: Enzym., 2013, 87, 30–36 CrossRef CAS PubMed.
- (a) K. Won, Y. H. Kim, E. S. An, Y. S. Lee and B. K. Song, Biomacromolecules, 2003, 5, 1–4 CrossRef PubMed; (b) H. Tonami, H. Uyama, S. Kobayashi, T. Fujita, Y. Taguchi and K. Osada, Biomacromolecules, 2000, 1, 149–151 CrossRef CAS PubMed.
- J. S. Dordick, M. A. Marletta and A. M. Klibanov, Biotechnol. Bioeng., 1987, 30, 31–36 CrossRef CAS PubMed.
- (a) T. Oguchi, S.-I. Tawaki, H. Uyama and S. Kobayashi, Macromol. Rapid Commun., 1999, 20, 401–403 CrossRef CAS; (b) H. Uyama, H. Kurioka, J. Sugihara and S. Kobayashi, Bull. Chem. Soc. Jpn., 1996, 69, 189–193 CrossRef CAS; (c) H. Uyama, H. Kurioka, I. Kaneko and S. Kobayashi, Chem. Lett., 1994, 423–426 CrossRef CAS; (d) J. A. Akkara, K. J. Senecal and D. L. Kaplan, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 1561–1574 CrossRef CAS PubMed.
- (a) H. Tonami, H. Uyama, S. Kobayashi and M. Kubota, Macromol. Chem. Phys., 1999, 200, 2365–2371 CrossRef CAS; (b) S. Dubey, D. Singh and R. A. Misra, Enzyme Microb. Technol., 1998, 23, 432–437 CrossRef CAS; (c) H. Uyama, H. Kurioka, J. Sugihara, I. Komatsu and S. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1453–1459 CrossRef CAS; (d) H. Uyama, H. Kurioka, J. Sugihara, I. Komatsu and S. Kobayashi, Bull. Chem. Soc. Jpn., 1995, 68, 3209–3214 CrossRef CAS; (e) N. Kommareddi, M. Tata, C. Karayigitoglu, V. John, G. McPherson, M. Herman, C. O'Connor, Y.-S. Lee, J. Akkara and D. Kaplan, Appl. Biochem. Biotechnol., 1995, 51–52, 241–252 CrossRef; (f) M. S. Ayyagari, K. A. Marx, S. K. Tripathy, J. A. Akkara and D. L. Kaplan, Macromolecules, 1995, 28, 5192–5197 CrossRef CAS; (g) A. M. Rao, V. T. John, R. D. Gonzalez, J. A. Akkara and D. L. Kaplan, Biotechnol. Bioeng., 1993, 41, 531–540 CrossRef CAS PubMed.
- R. Ikeda, N. Maruichi, H. Tonami, H. Tanaka, H. Uyama and S. Kobayashi, J. Macromol. Sci., Part A: Pure Appl.Chem., 2000, 37, 983–995 CrossRef PubMed.
- Y.-P. Xu, G.-L. Huang and Y.-T. Yu, Biotechnol. Bioeng., 1995, 47, 117–119 CrossRef CAS PubMed.
- (a) M. Akita, D. Tsutsumi, M. Kobayashi and H. Kise, Biosci., Biotechnol., Biochem., 2001, 65, 1581–1588 CrossRef CAS PubMed; (b) M. S. R. Ayyagari, D. L. Kaplan, S. Chatterjee, J. E. Walker and J. A. Akkara, Enzyme Microb. Technol., 2002, 30, 3–9 CrossRef CAS.
- A. N. P. Hiner, J. Hernández-Ruíz, M. B. Arnao, F. García-Cánovas and M. Acosta, Biotechnol. Bioeng., 1996, 50, 655–662 CrossRef CAS.
- T. Oguchi, S.-i. Tawaki, H. Uyama and S. Kobayashi, Bull. Chem. Soc. Jpn., 2000, 73, 1389–1396 CrossRef CAS.
- N. Mita, N. Maruichi, H. Tonami, R. Nagahata, S.-I. Tawaki, H. Uyama and S. Kobayashi, Bull. Chem. Soc. Jpn., 2003, 76, 375–379 CrossRef CAS.
- Y.-J. Kim, H. Uyama and S. Kobayashi, Macromolecules, 2003, 36, 5058–5060 CrossRef CAS.
- Y.-J. Kim, H. Uyama and S. Kobayashi, Macromol. Biosci., 2004, 4, 497–502 CrossRef CAS PubMed.
- Y.-J. Kim, K. Shibata, H. Uyama and S. Kobayashi, Polymer, 2008, 49, 4791–4795 CrossRef CAS PubMed.
- (a) S. S. Karajanagi, A. A. Vertegel, R. S. Kane and J. S. Dordick, Langmuir, 2004, 20, 11594–11599 CrossRef CAS PubMed; (b) J. M. Gómez, M. D. Romero and T. M. Fernández, Catal. Lett., 2005, 101, 275–278 CrossRef PubMed.
- Y. Peng, H. Liu, X. Zhang, Y. Li and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1627–1635 CrossRef CAS PubMed.
- N. Mita, S.-I. Tawaki, H. Uyam and S. Kobayashi, Macromol. Biosci., 2002, 2, 127–130 CrossRef CAS.
- H. Tonami, H. Uyama, S. Kobayashi, M. Reihmann and H. Ritter, e-Polym., 2002, 3, 1–7 Search PubMed.
- M. H. Reihmann and H. Ritter, Macromol. Chem. Phys., 2000, 201, 798–804 CrossRef CAS.
- R. Nakamura, Y. Matsushita, K. Umemoto, A. Usuki and K. Fukushima, Biomacromolecules, 2006, 7, 1929–1934 CrossRef CAS PubMed.
- Y. Pang, H. Ritter and M. Tabatabai, Macromolecules, 2003, 36, 7090–7093 CrossRef CAS.
- G. S. Nyanhongo, E. N. Prasetyo, E. H. Acero and G. M. Guebitz, Chem. Eng. Technol., 2012, 35, 1359–1372 CrossRef CAS PubMed.
- S. Sgalla, G. Fabrizi, S. Cacchi, A. Macone, A. Bonamore and A. Boffi, J. Mol. Catal. B: Enzym., 2007, 44, 144–148 CrossRef CAS PubMed.
- P. Zaragoza-Gasca, O. J. Villamizar-Gálvez, R. García-Arrazola, M. Gimeno and E. Bárzana, Polym. Adv. Technol., 2010, 21, 454–456 CAS.
- P. Zaragoza-Gasca, M. Gimeno, J. M. Hernández and E. Bárzana, J. Mol. Catal. B: Enzym., 2011, 72, 25–27 CrossRef CAS PubMed.
- H. Uyama, C. Lohavisavapanich, R. Ikeda and S. Kobayashi, Macromolecules, 1998, 31, 554–556 CrossRef CAS.
- H. Yoshizawa, J. Shan, S. Tanigawa and Y. Kitamura, Colloid Polym. Sci., 2004, 282, 583–588 CAS.
- A. Bilicia, I. Kayab and M. Yıldırımb, Des. Monomers Polym., 2011, 14, 353–366 CrossRef PubMed.
- S. Lv, D. Li, H. Ju, Y. Ma, C. Qiu and G. Zhang, J. Appl. Polym. Sci., 2013, 128, 523–529 CrossRef CAS PubMed.
- M. J. H. Van Haandel, M. M. J. Claassens, N. Van der Hout, M. G. Boersma, J. Vervoort and I. M. C. M. Rietjens, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1999, 1435, 22–29 CrossRef CAS.
- M. H. Reihmann and H. Ritter, J. Macromol. Sci., Part A: Pure Appl.Chem., 2002, 39, 1369–1382 CrossRef PubMed.
- R. Ghan, T. Shutava, A. Patel, V. T. John and Y. Lvov, Macromolecules, 2004, 37, 4519–4524 CrossRef CAS.
- S. Lv, R. Gong and Y. Ma, Polym. Adv. Technol., 2012, 23, 1343–1349 CrossRef CAS PubMed.
- S. Lv, R. Gong, X. Yan, M. Hou and G. Zhang, J. Appl. Polym. Sci., 2012, 125, 541–547 CrossRef CAS PubMed.
- (a) W. Schreiber, FEBS Lett., 1974, 41, 50–52 CrossRef CAS; (b) W. G. Rathmell and D. S. Bendall, Biochem. J., 1972, 127, 125–132 CAS.
- C. Goretzki and H. Ritter, Macromol. Chem. Phys., 1998, 199, 1019–1024 CrossRef CAS.
- L. Mejias, M. H. Reihmann, S. Sepulveda-Boza and H. Ritter, Macromol. Biosci., 2002, 2, 24–32 CrossRef CAS.
- F. F. Bruno, A. Trotta, S. Fossey, S. Nagarajan, R. Nagarajan, L. A. Samuelson and J. Kumar, J. Macromol. Sci., Part A: Pure Appl.Chem., 2010, 47, 1191–1196 CrossRef CAS.
- (a) N. Gochman and J. M. Schmitz, Clin. Chem., 1971, 17, 1154–1159 CAS; (b) N. Gochman and J. M. Schmitz, Clin. Chem., 1972, 18, 943–950 CAS.
- A. Kobayashi, Y. Koguchi, H. Kanzaki, S.-I. Kajiyama and K. Kawazu, Biosci., Biotechnol., Biochem., 1994, 58, 133–134 CrossRef CAS.
- H. Setala, A. Pajunen, I. Kilpelainen and G. Brunow, J. Chem. Soc., Perkin Trans. 1, 1994, 1163–1165 RSC.
- H. Setala, A. Pajunen, P. Rummakko, J. Sipila and G. Brunow, J. Chem. Soc., Perkin Trans. 1, 1999, 461–464 RSC.
- (a) M. Sottomayor, F. DiCosmo and A. R. Barceló, Enzyme Microb. Technol., 1997, 21, 543–549 CrossRef CAS; (b) A. E. Goodbody, T. Endo, J. Vukovic, J. P. Kutney, L. S. L. Choi and M. Misawa, Planta Med., 1988, 54, 136–140 CrossRef CAS PubMed.
- (a) E. Laurenti, E. Ghibaudi, S. Ardissone and R. P. Ferrari, J. Inorg. Biochem., 2003, 95, 171–176 CrossRef CAS; (b) E. Laurenti, E. Ghibaudi, G. Todaro and R. P. Ferrari, J. Inorg. Biochem., 2002, 92, 75–81 CrossRef CAS; (c) R. P. Ferrari, E. Laurenti and F. Trotta, J. Biol. Inorg. Chem., 1999, 4, 232–237 CrossRef CAS.
- (a) F. Saliu, E.-L. Tolppa, L. Zoia and M. Orlandi, Tetrahedron Lett., 2011, 52, 3856–3860 CrossRef CAS PubMed; (b) E. Bolzacchini, G. Brunow, S. Meinardi, M. Orlandi, B. Rindone, P. Rummakko and H. Setala, Tetrahedron Lett., 1998, 39, 3291–3294 CrossRef CAS.
- (a) D. Fournand, B. Cathala and C. Lapierre, Phytochemistry, 2003, 62, 139–146 CrossRef CAS; (b) K. Syrjanen and G. Brunow, J. Chem. Soc., Perkin Trans. 1, 1998, 3425–3430 RSC.
- (a) M. Bruschi, M. Orlandi, B. Rindone, P. Rummakko and L. Zoia, J. Phys. Org. Chem., 2006, 19, 592–596 CrossRef CAS PubMed; (b) M. Orlandi, B. Rindone, G. Molteni, P. Rummakko and G. Brunow, Tetrahedron, 2001, 57, 371–378 CrossRef CAS; (c) P. Rummakko, G. Brunow, M. Orlandi and B. Rindone, Synlett, 1999, 333–335 CrossRef CAS PubMed.
- D. Arrieta-Baez and R. E. Stark, Phytochemistry, 2006, 67, 743–753 CrossRef CAS PubMed.
- L. M. M. Mouterde, A. L. Flourat, M. M. M. Cannet, P.-H. Ducrot and F. Allais, Eur. J. Org. Chem., 2013, 173–179 CrossRef CAS PubMed.
- T. Takei, K. Sugihara, H. Ijima and K. Kawakami, J. Biosci. Bioeng., 2011, 112, 491–494 CrossRef CAS PubMed.
- D. N. A. Zaidel, I. S. Chronakis and A. S. Meyer, Food Hydrocolloids, 2012, 28, 130–140 CrossRef CAS PubMed.
- (a) K. Xu, F. Lee, S. J. Gao, J. E. Chung, H. Yano and M. Kurisawa, J. Controlled Release, 2013, 166, 203–210 CrossRef CAS PubMed; (b) K. M. Park, Y. Lee, J. Y. Son, D. H. Oh, J. S. Lee and K. D. Park, Biomacromolecules, 2012, 13, 604–611 CrossRef CAS PubMed; (c) F. Lee, J. E. Chung and M. Kurisawa, J. Controlled Release, 2009, 134, 186–193 CrossRef CAS PubMed.
- L. S. M. Teixeira, J. Feijen, C. A. van Blitterswijk, P. J. Dijkstra and M. Karperien, Biomaterials, 2012, 33, 1281–1290 CrossRef PubMed.
- B. Tang, Y. Wang, H. Liang, Z. Chen, X. He and H. Shen, Spectrochim. Acta, Part A, 2006, 63, 609–613 CrossRef PubMed.
- R. ter Haar, J. Wildschut, A. K. Sugih, W. Bart Möller, P. de Waard, C. G. Boeriu, H. J. Heeres, H. A. Schols and H. Gruppen, Carbohydr. Res., 2011, 346, 1005–1012 CrossRef CAS PubMed.
- G. Oudgenoeg, R. Hilhorst, S. R. Piersma, C. G. Boeriu, H. Gruppen, M. Hessing, A. G. J. Voragen and C. Laane, J. Agric. Food Chem., 2001, 49, 2503–2510 CrossRef CAS PubMed.
- G. Oudgenoeg, E. Dirksen, S. Ingemann, R. Hilhorst, H. Gruppen, C. G. Boeriu, S. R. Piersma, W. J. H. van Berkel, C. Laane and A. G. J. Voragen, J. Biol. Chem., 2002, 277, 21332–21340 CrossRef CAS PubMed.
- (a) K. Minamihata, M. Goto and N. Kamiya, Bioconjugate Chem., 2012, 23, 1600–1609 CrossRef CAS PubMed; (b) K. Minamihata, M. Goto and N. Kamiya, Bioconjugate Chem., 2011, 22, 2332–2338 CrossRef CAS PubMed; (c) K. Minamihata, M. Goto and N. Kamiya, Bioconjugate Chem., 2010, 22, 74–81 CrossRef PubMed.
- (a) A. Oosterveld, G. Beldman and A. G. J. Voragen, Carbohydr. Res., 2000, 328, 199–207 CrossRef CAS; (b) M. E. F. Schooneveld-Bergmans, M. J. W. Dignum, J. H. Grabber, G. Beldman and A. G. J. Voragen, Carbohydr. Polym., 1999, 38, 309–317 CrossRef CAS; (c) M. C. Figueroa-Espinoza and X. Rouau, Cereal Chem., 1998, 75, 259–265 CrossRef CAS; (d) A. Ng, R. N. Greenshields and K. W. Waldron, Carbohydr. Res., 1997, 303, 459–462 CrossRef CAS.
- T. Kiso, M. Shizuma, H. Murakami, T. Kiryu, K. Hozono, T. Terai and H. Nakano, J. Mol. Catal. B: Enzym., 2007, 45, 50–56 CrossRef CAS PubMed.
- E. Y. Dell, J. Consum. Health Internet, 2010, 14, 419–422 CrossRef.
- A. Wilkens, J. Paulsen, V. Wray and P. Winterhalter, J. Agric. Food Chem., 2010, 58, 6754–6761 Search PubMed.
- (a) Y. Takaya, K. Terashima, J. Ito, Y.-H. He, M. Tateoka, N. Yamaguchi and M. Niwa, Tetrahedron, 2005, 61, 10285–10290 CrossRef CAS PubMed; (b) Y. Takaya, K.-X. Yan, K. Terashima, Y.-H. He and M. Niwa, Tetrahedron, 2002, 58, 9265–9271 CrossRef CAS; (c) J. Ito and M. Niwa, Tetrahedron, 1996, 52, 9991–9998 CrossRef CAS.
- Y. H. He, Y. Takaya, K. Terashima and M. Niwa, Heterocycles, 2006, 68, 93–100 CrossRef CAS.
- C. Li, J. Lu, X. Xu, R. Hu and Y. Pan, Green Chem., 2012, 14, 3281–3284 RSC.
- I. K. Lee and B. S. Yun, J. Microbiol. Biotechnol., 2008, 18, 107–109 CAS.
- M. Sridhar, S. K. Vadivel and U. T. Bhalerao, Tetrahedron Lett., 1997, 38, 5695–5696 CrossRef CAS.
- S. Miyano, M. Tobita and H. Hashimoto, Bull. Chem. Soc. Jpn., 1981, 54, 3522–3526 CrossRef CAS.
- (a) A. I. Meyers and K. A. Lutomski, J. Am. Chem. Soc., 1982, 104, 879–881 CrossRef CAS; (b) J. M. Wilson and D. J. Cram, J. Am. Chem. Soc., 1982, 104, 881–884 CrossRef CAS.
- M. M. Schmitt, E. Schüler, M. Braun, D. Häring and P. Schreier, Tetrahedron Lett., 1998, 39, 2945–2946 CrossRef CAS.
- (a) A. Bodtke, W.-D. Pfeiffer, N. Ahrens and P. Langer, Tetrahedron, 2005, 61, 10926–10929 CrossRef CAS PubMed; (b) A. Bodtke, W.-D. Pfeiffer, N. Ahrens and P. Langer, Bioorg. Med. Chem. Lett., 2004, 14, 1509–1511 CrossRef CAS PubMed.
- D. Arrieta-Baez, R. Roman, R. Vazquez-Duhalt and M. Jiménez-Estrada, Phytochemistry, 2002, 60, 567–572 CrossRef CAS.
- (a) D. R. Buhler and H. S. Mason, Arch. Biochem. Biophys., 1961, 92, 424–437 CrossRef CAS; (b) H. S. Mason, I. Onopryenko and D. Buhler, Biochim. Biophys. Acta, 1957, 24, 225–226 CrossRef CAS.
- B. Halliwell and S. Ahluwalia, Biochem. J., 1976, 153, 513–518 CAS.
- J. S. Dordick, A. M. Klibanov and M. A. Marletta, Biochemistry, 1986, 25, 2946–2951 CrossRef CAS.
- A. M. Klibanov, Z. Berman and B. N. Alberti, J. Am. Chem. Soc., 1981, 103, 6263–6264 CrossRef CAS.
- H. Durliat, A. Courteix, M. Comtat and J.-L. Séris, J. Mol. Catal., 1992, 75, 357–369 CrossRef CAS.
- (a) A. Courteix and A. Bergel, Enzyme Microb. Technol., 1995, 17, 1087–1093 CrossRef CAS; (b) A. Courteix and A. Bergel, Enzyme Microb. Technol., 1995, 17, 1094–1100 CrossRef CAS.
- N. Bromberg and N. Durán, J. Mol. Catal. B: Enzym., 2001, 11, 463–467 CrossRef CAS.
- H. Krishna, P. Nagaraja, A. Shivakumar, K. Avinash and V. Lingaiah, J. Chin. Chem. Soc., 2013, 60, 452–459 CrossRef CAS PubMed.
- R. Ullrich and M. Hofrichter, Cell. Mol. Life Sci., 2007, 64, 271–293 CrossRef CAS PubMed.
- F. van Rantwijk and R. A. Sheldon, Curr. Opin. Biotechnol., 2000, 11, 554–564 CrossRef CAS.
- A. T. Smith and N. C. Veitch, Curr. Opin. Chem. Biol., 1998, 2, 269–278 CrossRef CAS.
- (a) S. P. de Visser, J. Phys. Chem. B, 2007, 111, 12299–12302 CrossRef CAS PubMed; (b) M. I. Savenkova, J. M. Kuo and P. R. O. de Montellano, Biochemistry, 1998, 37, 10828–10836 CrossRef CAS PubMed.
- R.-J. Dai, H. Huang, J. Chen, Y.-L. Deng and S.-Y. Xiao, Chin. J. Chem., 2007, 25, 1690–1694 CrossRef CAS PubMed.
- C. L. Budde, A. Beyer, I. Z. Munir, J. S. Dordick and Y. L. Khmelnitsky, J. Mol. Catal. B: Enzym., 2001, 15, 55–64 CrossRef CAS.
- Y. Sakihama, R. Tamaki, H. Shimoji, T. Ichiba, Y. Fukushi, S. Tahara and H. Yamasaki, FEBS Lett., 2003, 553, 377–380 CrossRef CAS.
- A. Sala, S. Nicolis, R. Roncone, L. Casella and E. Monzani, Eur. J. Biochem., 2004, 271, 2841–2852 CrossRef CAS PubMed.
- (a) E. Monzani, R. Rancone, M. Galliano, W. H. Koppenol and L. Casella, Eur. J. Biochem., 2004, 271, 895–906 CrossRef CAS PubMed; (b) J. B. Sampson, Y. Ye, H. Rosen and J. S. Beckman, Arch. Biochem. Biophys., 1998, 356, 207–213 CrossRef CAS PubMed.
- A. Pezzella, P. Manini, P. Di Donato, R. Boni, A. Napolitano, A. Palumbo and M. d'Ischia, Bioorg. Med. Chem., 2004, 12, 2927–2936 CrossRef CAS PubMed.
- (a) A. Palumbo, A. Napolitano, P. Barone and M. d'Ischia, Chem. Res. Toxicol., 1999, 12, 1213–1222 CrossRef CAS PubMed; (b) A. Napolitano, O. Crescenzi, A. Pezzella and G. Prota, J. Med. Chem., 1995, 38, 917–922 CrossRef CAS.
- P. Manini, E. Camera, M. Picardo, A. Napolitano and M. d'Ischia, Chem. Phys. Lipids, 2008, 151, 51–61 CrossRef CAS PubMed.
- D. Kumar, S. P. de Visser, P. K. Sharma, H. Hirao and S. Shaik, Biochemistry, 2005, 44, 8148–8158 CrossRef CAS PubMed.
- (a) S. Colonna, N. Gaggero, G. Carrea and P. Pasta, J. Chem. Soc., Chem. Commun., 1992, 357–358 RSC; (b) S. Colonna, N. Gaggero, A. Manfredi, L. Casella, M. Gullotti, G. Carrea and P. Pasta, Biochemistry, 1990, 29, 10465–10468 CrossRef CAS.
- S.-I. Ozaki and P. R. O. de Montellano, J. Am. Chem. Soc., 1994, 116, 4487–4488 CrossRef CAS.
- A. Tuynman, H. E. Schoemaker and R. Wever, Monatsh. Chem., 2000, 131, 687–695 CrossRef CAS.
- M. L. Ferrer, D. Levy, B. Gomez-Lor and M. Iglesias, J. Mol. Catal. B: Enzym., 2004, 27, 107–111 CrossRef CAS PubMed.
- Y. Xie, P. K. Das, J. M. M. Caaveiro and A. M. Klibanov, Biotechnol. Bioeng., 2002, 79, 105–111 CrossRef CAS PubMed.
- L. Galzigna, V. Rizzoli, M. P. Schiappelli, M. P. Rigobello, M. Scarpa and A. Rigo, Free Radical Biol. Med., 1996, 20, 807–811 CrossRef CAS.
- S. V. Dzyuba and A. M. Klibanov, Biotechnol. Lett., 2003, 25, 1961–1965 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |