
Rotating disk slurry electrodeposition of platinum at Y-zeolite/carbon Vulcan XC-72R for methanol oxidation in alkaline media
Amal Suleimana,
Christian L. Menéndeza,
Ramón Polancob,
Esteban Rosim Fachinic,
Yaritza Hernández-Lebróna,
Maxime J.-F. Guinelad,
Rolando Roque-Malherbe*b and
Carlos R. Cabrera*a
aDepartment of Chemistry and Molecular Sciences Research Building, University of Puerto Rico, 1390 Ponce De León Ave. Suite 2, San Juan, Puerto Rico 00926. E-mail: carlos.cabrera2@upr.edu
bInstitute for Physical Chemical Applied Research, School of Science, University of Turabo, PO Box 3030, Gurabo, Puerto Rico 00778-3030. E-mail: rroque@suagm.edu
cDepartment of Physical Sciences, General Studies College, University of Puerto Rico, Río Piedras Campus, San Juan, Puerto Rico 00931
dDepartment of Physics, University of Puerto Rico, San Juan, Puerto Rico 00931
First published on 17th December 2014
Abstract
Platinum was electrodeposited onto Y-zeolite and Y-zeolite (Y)/Vulcan XC-72R (V) to produce Pt/Y/V and Pt/Y catalysts using a rotating disk slurry electrode technique. The activities of the two catalysts were measured towards methanol electro-oxidation in alkaline media by cyclic voltammetry and chronoamperometry. The materials were examined using electron microscopy. The results were compared to those obtained on commercial catalysts. Pt/Y/V (with 14 wt% Pt) catalyst was the most active, even more so than a commercial Pt/V catalyst with 20 wt% Pt. The Pt/Y/V catalysts contained Pt nanoparticles and tetrahedra, likely a consequence of the nucleation and growth within the zeolite framework.
1. Introduction
Efforts are made to develop the direct electrochemical oxidation of alcohol and hydrocarbon fuels as an energy source.1–5 In particular, methanol has been largely investigated, since it is cheap, can be produced from fossil fuels or sustainable sources, and can be delivered the same way used today for petroleum.6 However, the commercial expansion of this fuel cell type depends largely on the catalysts since they are expensive and show unstability during long operation periods.7Direct methanol fuel cell (DMFC) is a variety of the polymer electrolyte fuel cell, conformed by a polymeric membrane that separates the anode and the cathode electrodes where methanol is used as a liquid fuel.8–11 The operation of the cell can produce catalyst loss by corrosion, catalyst particles sintering and catalyst poisoning.12 Therefore, having highly dispersed nanoparticles on conductive high surface area supports may improve the stability and robustness of the catalytic process. In this direction, efforts are made preparing highly dispersed catalysts on carbon based supports such as carbon black (carbon Vulcan XC-72R)13,14 carbon nanotubes15,16 ordered porous carbon,17 carbon nano-onions,14 nanodiamond,18 among others.7
Platinum, in general, is the most active catalysts.11 However, in order to efficiently use platinum as catalyst the metal must be highly dispersed in the form of small nanoparticles, since, with ultra-low loadings,4,7 therefore, reducing the total cost of the catalyst.19
Aluminosilicate zeolites supports have been used in methanol and CO oxidation as well as for the water–gas-shift reaction.20–22 Their structure, composition and properties that offer an elevated ionic strength environment. Rolison has shown the use of these materials in electrochemistry.23–25 The properties of aluminosilicate zeolites responsible for affecting charge transfer reactions in electrochemical systems23 are, the zeolite size and shape selectivity due to their rigid pores and channels, the ion exchange properties, and their catalytic properties. In this regard, hydrated zeolites conduct through intracrystalline and intercrystalline cationic conduction, given that, the material behave as an electrolyte, by the water adsorbed in the primary and secondary porosity of the zeolite.26 On this ground, in the formulation of the electro-catalyst included in the DMFC is possible to incorporate an additional component, namely an aluminosilicate zeolite,27 since this material in an aqueous electrolyte solution may work as a solid ion conductor,28 provide an adsorption space29 and supply ions by ionic exchange.30,31
The tested metallic catalysts were produced by electrochemical reduction of platinum from K2PtCl6on the plain zeolite and a zeolite–carbon black composite, using the rotating disk-slurry electrode (RoSDE) technique. The produced catalysts were compared with three standard commercial catalysts, specifically, Pt-black, 20 wt% Pt/Vulcan XC-72R (ETEK) and 40 wt% Pt/Vulcan XC-72R (ETEK) catalysts. The materials were examined using scanning and transmission electron microscopy (SEM, TEM), thermo-gravimetric analysis (TGA), powder X-ray diffraction (PXRD), inductive couple plasma-optical emission spectrometry (ICP-OES), X-ray photoelectron spectrometry (XPS), diffuse reflectance Fourier transform infrared spectrometry (DRIFTS), and carbon dioxide adsorption. Finally, the electrochemical measurements were done for methanol oxidation in alkaline media.
2. Experimental method
2.1 Materials
All the chemicals were analytical grade without additional purification. The water used in the synthesis process was bi-distilled. Vulcan XC-72R (Cabot) and zeolite Na–Y (Si/Al = 2.4) (Sigma-Aldrich) were used as support materials. The Vulcan was sonicated in 1.0 M H2SO4 for 8 h previous to be used to increase the oxygen in the surface of the carbon black particles and disperse it in the slurry. The zeolite was treated for the dealumination process and to exchange Na+ by H+ ions.30,33,34The electro-deposition of the platinum nanoparticles was performed using the rotating disk-slurry electrode (RoDSE) methodology, following a procedure previously developed in a three compartments, separated by fritted glass, electrochemical cell. The cell was composed of a rotating disk electrode (RDE) (PINE Instrument Company) in the central section, a graphite rod, was used as counter electrode, and a Ag/AgCl (+0.197 V versus NHE) (PINE Instrument Company) was the reference electrode, both in aqueous sulfuric acid solution (0.1 M), included in the right and left sectors of the cell respectively. The disk electrode was a glassy carbon electrode (Bioanalytical System) with an area of 0.20 cm2. The commercial catalysts used to compare the activity of the Pt/Y/V catalysts were Pt black (Johnson Matthey, high surface area), 20 wt% Pt/Vulcan XC-72R (ETEK) and 40 wt% Pt/Vulcan XC-72R (ETEK).
2.2 Catalyst preparation using the RoDSE electrodeposition technique
The procedure used for the preparation of the Pt/Y/Vulcan (Pt/Y/V) and Pt/Y catalysts was used as reported by Santiago et al.13 Fig. 1 shows a scheme of RoDSE process where metal precursor is dissolved into dispersed in a slurry solution of a colloidal support, and an electrodeposition potential is applied to the RDE while rotating at 900 rpm. Briefly, a concentrated suspension was formed with 50 mg of zeolite in 20 mL of 0.1 M H2SO4, this slurry was placed in the center of the three-electrode cell assembly for the platinum electro-deposition. Then 5.00 mL of the 3 mM K2PtCl6 (Aldrich) was added to the suspension in the center compartment to make the slurry solution. The electrochemical cell was sealed, and purged with nitrogen for 1 h, while the RDE was rotated at 900 rpm in the slurry. The electro-deposition took place at a constant potential, −0.200 V vs. Ag/AgCl (Basic Electrochemical System of EG&G) for 2 h. The electro-deposition was repeated six additional times (i.e. 6 × 2 h) while adding the platinum complex solution (5.00 mL of 3 mM K2PtCl6). Fig. 1 shows an illustration of the electrodeposition technique describe above. Afterwards, the slurry was filtered and rinsed with 600 mL of nanopure deionized water (18.2 MΩ cm NANOpure Diamond of Barnstead) to remove the impurities. The filtered sample was air-dried. For the Pt/Y/V catalyst preparation, slurry of 50 mg of Vulcan–zeolite (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in 20 mL of 0.1 M H2SO4 was placed in the center of the three-electrode cell assembly for the platinum electro-deposition. Afterwards, the methodology was repeated for the preparation of the Pt/Y sample.
:1) in 20 mL of 0.1 M H2SO4 was placed in the center of the three-electrode cell assembly for the platinum electro-deposition. Afterwards, the methodology was repeated for the preparation of the Pt/Y sample.
 | ||
| Fig. 1 Schematic illustration of the RoDSE technique process. | ||
2.3 Preparation of catalyst coated glassy carbon (GC) electrodes
To produce the ink-paste-electrode (IPE), a suspension containing 1.0 mg of the catalytic nanomaterial, 100 μL of nano-pure water and 200 μL of isopropanol was placed under ultrasound for 30 minutes. Then, 8 μL of Fumion® (5% w/w polyarylene sulfonic acid polyelectrolyte dissolved in water/isopropanol, FuMA-Tech GmbH, St. Ingbert, Germany) was added and stirred for 30 minutes. The electrode was prepared by adding 8 μL of the ink paste to the glassy carbon electrode (3 mm diameter, Bioanalytical System) and then air dried for 30 minutes.2.4 Characterization methods
The samples were examined using two scanning electron microscopes (SEM, JEOL, JSM-7500F and JSM-6480LV). The catalysts were also investigated using a high resolution transmission electron microscope (HRTEM, JEOL, JEM-2200FS). The elemental composition of the Pt/Y/Vulcan XC-72R catalyst was measured using Perkin Elmer Inductively Coupled Plasma-Optical Emission Spectrometer (ICP-OES) Optima 8000.X-ray photoelectron spectroscopy (XPS) (PHI 5600ci) surface analysis was done using an Al Kα monochromatic X-ray source (350 W) and a hemispherical electron energy analyzer. The analysis was made pressing the catalytic powder onto a Mo film.
Powder X-ray diffraction (PXRD) (Bruker D8 Advance) profiles were collected using a Bragg–Brentano vertical goniometer configuration. The thermal gravimetric analysis (TGA) (TA, Q-500) measurements were done from 25 to 700 °C, at a heating rate of 5 °C min−1, while flowing 100 mL min−1 of pure N2. Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was done by using a Thermo Scientific Nicolet iS10 FTIR spectrometer. The data were collected at a resolution of 4 cm−1 employing 100 scans per sample. A background, with pure KBr, provided by Nicolet, using the same conditions was always made prior to sample analysis. The hydrated samples spectra were obtained at room temperature under N2 flow (Praxair, 99.99%), at 50 mL min−1 rate. The spectra of the dehydrated samples was done by heating at 473 K under a 50 mL min−1 flow of N2 (Praxair, 99.99%) for 2 hours. The spectra of the degassed materials were subsequently obtained at room temperature, under N2 flow. Additionally, DRIFTS spectra of carbon dioxide (Praxair, 99.99%) adsorbed in the Pt/Y/V catalyst were obtained utilizing as background the dehydrated sample at room temperature. After that, CO2 (Praxair, 99.99%) flow at a 50 mL min−1 rate, for three minutes, was passed through the dehydrated samples, afterward, the sample was purged under N2 (Praxair, 99.99%) flow at a rate of 50 mL min−1 for one minute, next, a spectrum of the carbon dioxide molecule adsorbed on the catalyst was obtained at room temperature under N2 flow.
Carbon dioxide (Praxair, 99.99%) adsorption19 at 273 K and pressure up to ca. 1 atm. on samples degassed at 473 K for three hours under high vacuum (10−6 Torr), was carried out using an upgraded Quantachrome Autosorb-1 automatic volumetric physisorption analyzer. The backfilling process was carried out using helium (Praxair, 99.99%).
2.5 Cyclic voltammetry and constant potential experiments
The electrochemical activity of the catalysts were measured using a potentiostat/galvanostat system (EG&G) and a three electrode compartment electrochemical cell, separated by glass frits.13,14 The potentials measured in this study were referenced to Ag/AgCl. The cyclic voltammetric (CV) experiments of the tested catalysts were done in 0.1 M KOH using a potential range of between −0.9 and 0.6 V vs. Ag/AgCl and a potential sweep rate of 100 mV s−1 at room temperature. The counter electrode was a Pt wire. All electrolyte solutions were bubbled with argon gas for 10 min before use. The Pt active surface areas were calculated from the cyclic voltammograms in 0.1 M KOH solutions using the hydrogen adsorption–desorption region. In addition, the activity of the Pt/Y/V catalyst towards methanol oxidation in basic media, 1.0 M methanol and 0.1 M KOH, was done and compared against that of Pt black and 40% Pt/Vulcan XC-72R (ETEK) catalysts.The onset potential for each catalyst was determined at 5% of the maximum anodic peak current density for the methanol oxidation reaction. The chronoamperometry experiment was done with an applied potential of −0.4 V vs. Ag/AgCl in 1.0 M methanol and 0.1 M KOH aqueous solution at room temperature.
2.6 CO stripping experiment
To perform CO oxidative stripping experiments in alkaline medium, the ink paste electrodes were placed in a solution of 0.1 M KOH. Then, CO adsorption on the Pt surface was done by bubbling high purity CO through the working electrode cell compartment for 1200 seconds at a constant applied potential of −0.7 V vs. Ag/AgCl. Afterward, N2 was bubbled for 300 seconds at the same potential to remove any remaining CO present in solution. The final step was a linear sweep voltammetry (LSV) from −0.7 V to 0.7 V vs. Ag/AgCl at a sweep rate of 20 mV s−1.3. Results and discussion
3.1 Morphology and structure of the catalysts
The structure and chemical identity of Pt/Y/V were verified by the SEM, HRTEM and XRD studies. All the SEM images present nearly cubic zeolite crystals.30 In Fig. 2, the platinum nanoparticles appear brighter. At the same time as the SEM study, an EDAX elemental analysis of the as-synthesized catalysts was done in five different spots. The weight percent (wt%) average values were 27% platinum, 12% oxygen, 7.6% silicon and 53% carbon. | ||
| Fig. 2 Scanning electron microscopy images of the Pt/Y/V catalysts prepared by RoDSE. | ||
The structure and chemical identity of Pt/Y/V were verified by HRTEM and XRD studies. The prepared sample by RoDSE technique was characterized by high resolution transmission electron microscope to detect if the platinum were deposited onto the carbon/zeolite support. High resolution TEM images showing platinum nanostructures are displayed in Fig. 3. The nanoparticles measured in HRTEM had a particle size of 4 to 9 nm. Fig. 3A and B shows Pt nanoparticles. This is likely a result of the nucleation of Pt on the carbon Vulcan XC-72R. Fig. 3C and D show Pt tetrahedron nanostructures. This is likely a result of the nucleation of Pt in the framework of the zeolite. XRD data suggested that there were 8 (1) nm nanoparticles of Pt. The XRD pattern (Fig. 4) shows the face-centered cubic crystal structure of Pt with the presence of (111), (200), (220), (311) and (222) planes, demonstrating highly polycrystalline structure.
 | ||
| Fig. 3 TEM images showing the Pt crystallites in the Y/V catalyst. | ||
 | ||
| Fig. 4 X-ray diffraction of the Pt/Y/V sample prepared by RoDSE. | ||
The contribution of an addition of Vulcan XC-72R to Pt/HY was the ability to carry or conduct an electrical current and to avoid segregation or agglomeration of Pt. The presence and distribution of platinum, Vulcan and zeolite in this catalyst is confirmed by EDX elemental mapping analysis shown in Fig. 5.
 | ||
| Fig. 5 (a) Field emission electron microscopy images of the Pt/Y/V catalyst on a carbon tape. EDX elemental mapping of (b) carbon (red), (c) oxygen (green), (d) sodium (dark blue), (e) aluminum (yellow), (f) silica (pink), (g) platinum (cyan) of the catalyst in (a). (h) Combined mapping image of both C and Pt. | ||
Fig. 6a presents X-ray photoelectron spectroscopy (XPS) spectrum of the Pt 4f binding energy region corresponding to the as-synthesized Pt/Y/V.35 Fig. 6b and d are exhibited the O 1s36 and C 1s37 binding energy regions of the XPS profiles. Further, in Fig. 6d is displayed the XPS peak corresponding to the Si 2p.
 | ||
| Fig. 6 X-ray photoelectron spectroscopy profiles corresponding to the as-synthesized Pt/Y/V catalyst. | ||
XPS showed a composition of 17.0% platinum, 9.11% oxygen, 4.17% silicon, 69.25% carbon and 1.0% chlorine on the surface of sample.
High resolution XPS spectra for the Pt 4f binding energy peak were analyzed to identify the platinum oxidation states on the prepared sample by the RoDSE technique. The XPS binding energy data revealed that the majority of the platinum was in its metallic form26,27 in the Pt/Y/V catalyst (Fig. 6a), i.e., the RoDSE Pt electrodeposition process was highly efficient. Fig. 6a shows the curve-fitting analysis for the Pt/Y/V sample, which appear to have two different platinum species, Pt(0) and Pt(II). The first set of XPS doublets in the binding energy of 71.3 (55.46% area) and 74.6 (41.6% area) eV is the most intense and correspond to Pt(0) followed by Pt(II) which is less intense and corresponds is found at 74.5 (1.69% area) and 77.9 (1.27% area) eV. The appearances of these two platinum species were as expected because of the use of Pt complex as a precursor. On the other hand, the peak in the C 1s binding energy region correspond to graphitic carbon.37
In Fig. 7 are exhibited the XRD profiles of the Pt/Y/V and commercial catalysts samples at room temperature. These XRD profiles were resolved into separate Bragg components using the Pawley whole-powder pattern decomposition method while the cell parameter was refined38 knowing that platinum crystallizes in the cubic Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. Besides, the Gaussian crystallite size (φPt) was calculated.39–41
m space group. Besides, the Gaussian crystallite size (φPt) was calculated.39–41
 | ||
| Fig. 7 X-ray diffraction profiles of the Pt/Y/V, Vulcan XC-72R, NAY zeolite, Pt black, 20% Pt/Vulcan XC-72R and 40% Pt/Vulcan XC-72R. | ||
The computer program used to perform these calculations was the Bruker DIFFRACplus TOPAS™ software. The calculated Gaussian crystallite size is 5(1) nm 40 wt% Pt/Vulcan XC-72R, 4(1) nm 20 wt% Pt/Vulcan XC-72R, 7(1) nm platinum black and 8(1) nm Pt/Y/V. Subsequently, to verify the loss of the guest molecules (fundamentally H2O) throughout heating and study further structural features, the thermal gravimetric analysis (TGA) method was used (Fig. 8).38
 | ||
| Fig. 8 Thermal gravimetric analysis (TGA) profiles (a) and TGA derivative profiles of Pt/Y/V (b), Pt/Y (c) and Y zeolite (d). | ||
The TGA profile of the Pt/Y/V and Pt/Y catalysts and the Na–Y zeolite are exhibited in Fig. 8a. Meanwhile, in Fig. 8b–d are reported the TG profile derivatives of both catalysts and the zeolite. These results indicated that adsorbed water was released from ca. 50 to 200 °C. Afterwards, zeolite dehydroxylation from ca. 200 to 700 °C was produced,42 next, was evidenced the characteristic slight decrease of weight from 200 to 700 °C, normally observed in carbon black.43 Further, with the TGA data were calculated the amount of platinum contained in the Pt/Y/V and Pt/Y catalysts, by the comparison of the water weight lost by the pure zeolite Y, the Y/Pt/V, and the Pt/Y catalysts, namely, 0.24 g g−1, 0.08 g g−1 and 0.23 g g−1. With this information was estimated that, ca. 0.40 ± 0.03 g g−1 of zeolite Y and 0.40 ± 0.03 g g−1 of Vulcan were contained in the Pt/Y/V catalyst, since by design the proportion of zeolite Y and Vulcan was 1:1. On this ground, the amount of platinum in the Pt/Y/V catalyst was, 0.19 ± 0.06 g g−1, while the amount of Pt in the Pt/Y catalyst was, 0.1 ± 0.02 g g−1. The ICP data showed the amount of Pt 14.7%, Si 32.2%, Na 0.59%, Al 0.61%, and the remaining 51.9% was carbon, oxygen, and nitrogen. From the ICP data we see a slightly lower value than the TGA data. Nevertheless, the ICP data confirms the dealuminization process when placing the NaY in 0.1 M H2SO4 and that the Pt electrodeposition is possible at NaY/Vulcan using the RoDSE technique. Finally, the small amount of Na is indicative that an ion exchange with H+ and Pt complex during the sonication and electrodeposition process. Consequently, the zeolite include in the catalyst is in the acid form that is HY zeolite. In the presence of Pt complex in the acid solution may have had an intercalation process in the zeolite framework leading to the Pt tetrahedral formation (see Fig. 3C and D).
An estimation of the chemical surface area (SCSA) of the platinum phase included in the produced, Pt/Y/V, Pt black and the commercial catalysts ETEK (40% Pt/Vulcan XC-72R) was made, using the following relation, SCSA = 6/Φρ. In this regard knowing that the measured crystallite size was, 4 to 9 nm for the Pt/Y/V catalysts respectively and the platinum density, ρ = 21.5 g cm−3, then is possible the estimation of SCSA for the produced catalysts. The chemical surface area of Pt/Y/V is between 30 m2 g−1 to 70 m2 g−1. Besides, using data previously published,44 namely, SCSA for the 40% Pt/Vulcan XC-72R ETEK was 111 m2 g−1, whereas, the specific surface area of the Pt black was 28 m2 g−1.45
Later, the DRIFTS spectra of the hydrated (as-synthesized) and dehydrated Pt/Y/V in the range between: 600–3900 cm−1 were collected (Fig. 9a). The main IR active vibrations observed can be classified in two main regions. In the first expanse, from 600 to 2000 cm−1, appear bands corresponding to the zeolite structure, meanwhile the second section of the spectrum, namely, between 3200–3700 cm−1, display the stretching, ν(OH) vibration produced by adsorbed water.31 Further, the DRIFTS spectrum of the Pt/Y/V catalyst, dehydrated at 200 °C in N2 flow, is exhibited in Fig. 9b. This spectrum exhibit a peak at ca. 3605 cm−1 related with bridging acid hydroxyl (Si–OH–Al) groups, and displays a very small band at ca. 3700 cm−1, corresponding to the silanol (Si–OH) groups.44 The acid sites were produced by the electro-deposition, since this process was performed in acid media, therefore, as was previously stated, the Na+ ions included in the Na–Y zeolite were exchanged by H+, producing the detected bridging acid hydroxyl (Si–OH–Al) groups. Furthermore, the DRIFTS spectrum of carbon dioxide, adsorbed at 760 Torr on the dehydrated Pt/Y/V can be examined in Fig. 10.
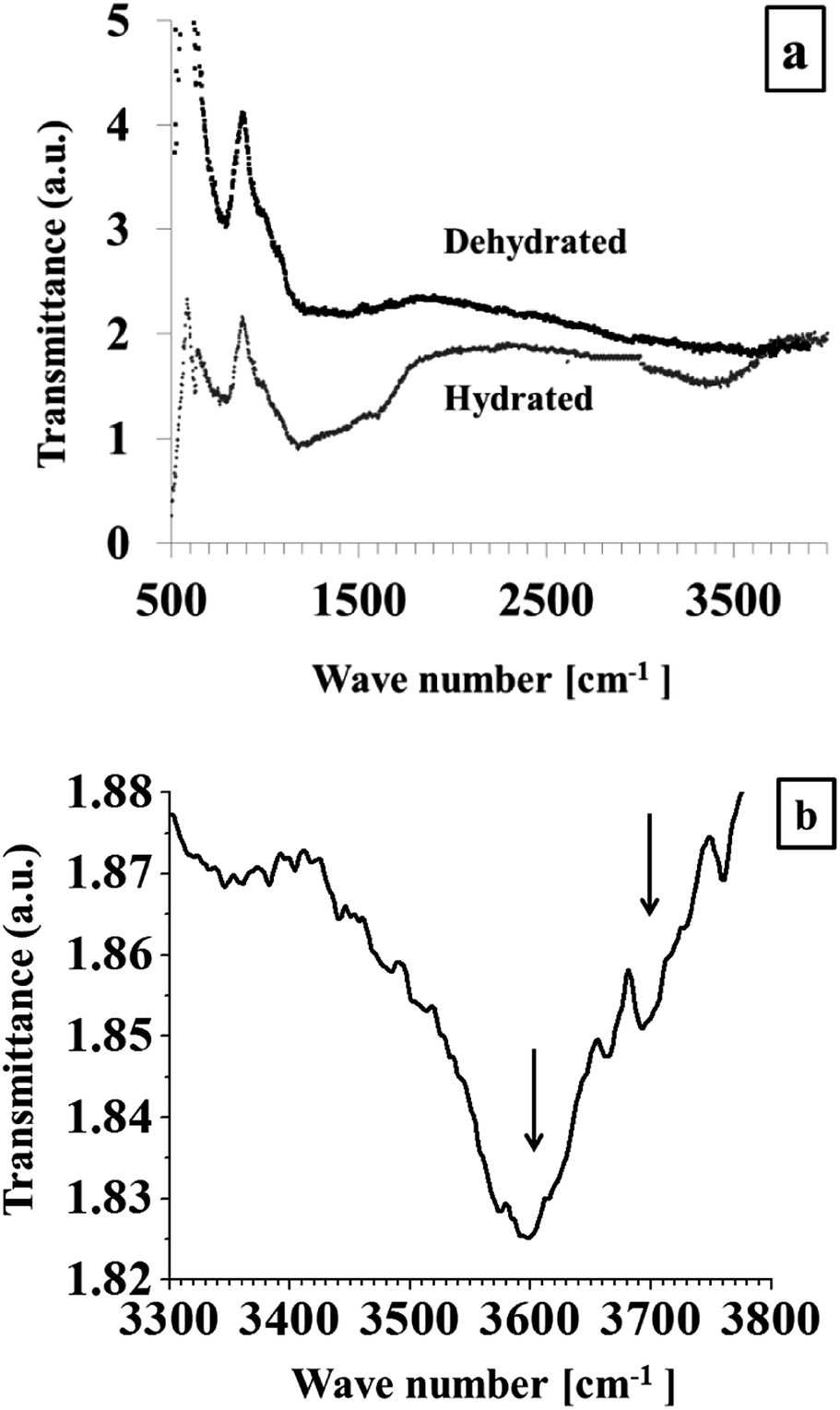 | ||
| Fig. 9 DRIFTS spectra of the hydrated and dehydrated Pt/Y/V catalyst (a), and spectrum in the OH region (b). | ||
 | ||
| Fig. 10 DRIFTS spectrum of adsorbed carbon dioxide on the dehydrated zeolite contained in the Pt/Y/V catalyst. | ||
The free carbon dioxide molecule belongs to the D∞h group of symmetry, exhibiting four fundamental vibration modes, namely, the symmetric stretching, ν1 (1338 cm−1), the doubly degenerate bending vibration, ν2a and ν2b (667 cm−1), and the asymmetric stretching vibration ν3 (2349 cm−1).46 The, ν2 and ν3, modes are infrared active, whereas ν1 is only Raman active in the free molecule.47 The asymmetric stretching vibration, ν3, of the adsorbed carbon dioxide molecule, at ca. 2332 cm−1 corresponds to carbon dioxide physically adsorbed inside the Y zeolite framework.48 A red shift is observed the molecule is adsorbed. Note that 2349 is larger than 2332, indicating that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonding is weaker when the molecule is adsorbed.
O bonding is weaker when the molecule is adsorbed.
As a final point of the present section was performed the fitting of the Dubinin–Radushkevitch (DR) adsorption isotherm equation,49 ln(na) = ln(Na) − (RT/βE)2[ln(P0/P)]2, where, E, is a parameter known as the characteristic energy of adsorption, β, is the affinity coefficient, and, Na, is the maximum amount adsorbed in the micropore volume, to the experimentally measured adsorption data, specifically, the amount adsorbed, na, at 273 K, of CO2 on the Pt/V/Y catalyst and the inverse of the relative pressure, P0/P, in the range, 0.002 < P/P0 < 0.03.23 The process allowed the calculation of the best fitting parameters, namely, Na and E. After that, the micropore volume was computed using the Gurvich rule,19 i.e., WMP = NaVL where VL = 48.3 cm3 g−1 is the carbon dioxide molar volume, yielding, WCOMP2 = 0.105 cm3 g−1. Then, since the micropore volume of pure zeolite Y is ca. 0.30 cm3 g−1, subsequently, the amount of zeolite Y in the Pt/V/Y catalyst is ca. 0.33 ± 0.08 g g−1,48 a quantity consistent with the TGA data.
3.2 KOH cyclic voltammetry and surface area determination
To properly assess the activity of an electro-catalyst, the currents produced as a result of the electrochemical reaction must be normalized, namely, calculated with respect to the real surface area in our case the electrochemical surface area (ECSA).50 In this regard, in Fig. 11 are reported the normalized cyclic-voltammetry curves corresponding to Pt/Y/V, NaY, Vulcan XC-72R, 40% Pt-Vulcan XC-72R (ETEK) and Pt black catalysts in 0.1 M KOH. | ||
| Fig. 11 Cyclic voltammetry curves of Pt/Y/V, NaY, Vulcan XC-72R, 40% Pt-Vulcan XC-72R (ETEK) and Pt black catalysts in 0.1 M KOH. | ||
The following reaction,51,52 Pt + H+ + e− ↔ Pt − Had, describes hydrogen adsorption–desorption, in basic media, in the cathodic and anodic sections of the voltammograms. The characteristic hydrogen adsorption–desorption phenomena are manifested as peaks in the voltammograms, next, is found the double-layer effect, and, afterwards, the oxide formation peak is perceived. In the reported voltammograms evidence these features. Particularly, the hydrogen adsorption–desorption phenomena are notably informative, given that, platinum nanoparticles exhibit certain crystallographic planes at the surface, known as facets, displaying the (111), (100), and (110) indexes.53–55 The presence of these facets induces in the voltammograms peaks location at different potentials. The principal shapes encountered for Pt nanoparticles are, tetrahedral–octahedral exhibiting the (111) and (110) basal planes, cubic exhibiting the (100), hexagonal exhibiting the (100) and (111) and near spherical nanoparticles exhibiting the (111) and (100) facets.53,54 Hence, is possible to affirm that the peaks reported in Fig. 9 are related to polycrystalline nanoparticles,56 given that, platinum nanoparticles normally tend to display a combination of (111) and (100) facets to reduce the particle interfacial free energy.
Meanwhile, the distinctive hydrogen adsorption–desorption phenomena in the Pt/Y catalyst were not clearly displayed as peaks in the voltammograms. This fact indicates that plain zeolite is an unsuitable support to produce an electro-catalyst. In other words, the presence of a conducting support is, in general, necessary in the catalyst formulation. Moreover, the capacitance was higher in the all catalysts with zeolites.
3.3 Methanol electrooxidation
The activity of Pt/Y/V, ETEK and Pt-black towards methanol oxidation in basic media: CH3OH + 6OH− → CO2 + 5H2O + 6e−, was investigated by cyclic voltammetry in degassed CH3OH (1 M)–KOH (0.1 M) aqueous solutions (Fig. 12 and Table 1). | ||
| Fig. 12 Cyclic-voltammetry curves of Pt/Y/V, Pt black, and ETEK (40% Pt-Vulcan XC-72R) catalysts in degassed CH3OH (1 M)–KOH (0.1 M) aqueous solutions. | ||
| Catalyst |
|
At maximum peak current potential [V versus Ag/AgCl] |
|---|---|---|
| Pt/Y/V | 1.72 | 0.00 |
| Pt-black | 1.10 | −0.04 |
| ETEK | 0.34 | −0.02 |

The onset potential (Es) for methanol oxidation is at −0.35 V vs. Ag/AgCl.
The maximum current densities of the three catalysts are 2.5 (Pt/Y/V), 1.7 (40% Pt/Vulcan XC-72R), and 0.5 mA cm−2 of Pt (Pt black), respectively.
Fig. 13 shows the chronoamperometric experiments which was carried out to observe the stability and possible poisoning between the ETEK and Pt/Y/V under short time continuous operation toward methanol oxidation reaction. To measure the tolerance of the Pt/Y/V nanocatalysts to methanol oxidation reaction, it conducted chronoamperometry tests in 1.0 M CH3OH/0.1 M KOH for 1800 s. As can been seen from Fig. 10, the potentiostatic current decreases rapidly in the initial period of time; and this can be due to the formation of COads and other intermediate species, such as CH3OHads, CHOads and OHads, during methanol oxidation reaction. The Pt/Y/V seems to be clearly more stable because exhibit a tolerance for intermediate species formed during the methanol oxidation than the commercial catalysts.
 | ||
| Fig. 13 Chronoamperometry of Pt/Y/V, 40% Pt/Vulcan XC-72R, and Pt black in 1.0 M CH3OH/0.1 M KOH at an applied potential of −0.4 V vs. Ag/AgCl. | ||
3.4 CO stripping experiment
One of the principle facts to take in consideration at the time of doing characterization of the nanocatalysts material for fuel cells is the resistance of the CO poisoning. CO stripping voltammetry is commonly used to test the activity of a catalyst for electrochemically oxidizing adsorbed CO on the catalyst. In Fig. 14 shows the CO stripping voltammograms of the Pt/Y/Vulcan and 40% Pt/Vulcan XC-72R catalysts in 0.1 M KOH at 25 °C after full adsorption of CO and subsequent purging of the solution with high-purity argon at a scan rate of 25 mV s−1. CO stripping voltammetry is a technique used to calculate the Pt–CO saturated coverage at Pt. The charge required to oxidize the saturated CO layer on a NaY/Vulcan/Pt was determined from the electrochemical CO desorption method and assuming a full CO–Pt coverage, which is used for Pt surface area determination. Basically, the CO adsorption method is the same as the hydrogen adsorption method i.e. a probe molecule is adsorbed at the Pt surface (at potential where CO oxidation does not occur). Pt catalysts supported on zeolite have been reported for the removal of CO, taking advantage of its “chemical and/or physical molecular sieve effect” to make CO react with oxygen.23 In Fig. 14 we see a lower CO stripping potential for Pt/Y/V than for 40% Pt/Vulcan XC-72R. | ||
| Fig. 14 CO stripping voltammograms of the NaY/Vulcan/Pt and 40% Pt/Vulcan XC-72R catalysts in 0.1 M KOH at 25 °C after full adsorption of CO and subsequent purging of the solution with high-purity argon at a scan rate of 25 mV s−1. | ||
Another way to evaluate the electro-catalytic activity for methanol oxidation, the turnover number (TON) is normally used (Table 1).57
The TON is just the number of revolutions of the catalytic cycle per second, consequently is a parameter showing effects close to the atomic-molecular nature of the catalytic process. This parameter, in the concrete case of electro-catalytic methanol oxidation, is defined as the number of reacted methanol molecules per metallic catalytic site, in the catalyst surface within one second. The turnover number is estimated, using the maximum oxidation peak current in the cyclic voltammogram, with the following relation:5,58
 | (1) |
In the complex process produced during methanol oxidation, the extent of the anode peak current and the TON characterize the electro-catalytic activity of the catalyst toward methanol oxidation. In this regard, the Pt/Y/V catalyst exhibited current density and TON values higher for methanol oxidation than that of the Pt black and ETEK catalysts, although the surface area of the platinum black and ETEK catalysts are higher than that of the Pt/Y/V catalyst (Table 1).
3.5 Role of the zeolite included in the catalyst in the reaction mechanism
In the methanol oxidation electrochemical reaction many electrons are released, therefore, this whole process requires a complicated mechanism involving several steps, namely, adsorbed intermediates and side-products to be produced, then, if some of these intermediates are irreversibly adsorbed (e.g. CO) on the platinum catalyst the course of the reaction is hindered. In this regard, in both acid and basic media the oxidation of adsorbed CO may occur via the Langmuir–Hinshelwood mechanism:60| COad + OHad → CO2 + H+ + e− | (2) |
The main result evidenced by Fig. 12 and Table 1 is that the Pt/Y/V catalyst presented a high activity towards methanol oxidation when compared to Pt black and ETEK. This fact should be related to this catalyst lower CO poisoning. To explain this finding is necessary to take into account that Y zeolite is an excellent adsorbent in gaseous and as well in liquid phase,61 therefore, is reasonable to suppose that the mechanism producing the high activity of the Pt/V/Y catalyst is CO2 adsorption by the zeolite phase included in the catalyst formulation, given that, the removal of this molecule promote the oxidation of adsorbed CO (eqn (2)). However, carbon dioxide interacts with water, according to the following equilibrium reaction CO2 + H2O ↔ HCO3− + H+, on this ground, the zeolite should adsorb solvated molecular carbon dioxide and bicarbonate anions.
The framework of zeolite Y is composed of sodalite cages, linked by 6-6 secondary building units, located in a face centered cubic lattice, where half of the tetrahedral sites are occupied by sodalite cages as is the case in the structure of diamond.30,31 This framework, known as FAU type, contains 12-member rings (MR) windows which lead to approximately spherical cavities, with a radius of R = 6.9 Å, known as β cages, that are connected in tetrahedral symmetry, and opened though four 12-member rings (MR) windows each one with a diameter of d = 7.4 Å.62 When the solvated carbon dioxide and bicarbonate molecules are immersed in the Y zeolite adsorption space31 they become subjected to diverse interaction fields, such as, the dispersion, repulsion energies, the electrostatic interactions, namely, polarization, field dipole and field gradient quadrupole interactions48 and the adsorbate–adsorbate interaction.63 On this ground, the structure and surface chemistry of the H–Y zeolite, included in the catalyst, allows carbon dioxide adsorption in liquid phase. This fact explains why the Pt/Y/V catalyst is more active than both the Pt black and ETEK catalysts.
In general, to reduce the CO poisoning problem the platinum catalyst is alloyed with Ru, Sn, W, Mo, and other metals oxides to produce bifunctional sites in the catalyst surface.32 On the surface of this alloy, the Pt sites are centers for CO adsorption and dehydrogenation, while on the other metallic sites present in the alloy nanoparticles surface, the OHad adsorbed species react with the adsorbed COad to produce, CO2, in consequence, the CO poisoning species is detached from the catalyst.64 Platinum–ruthenium alloys nanoparticles are considered the most active electro-catalysts for methanol oxidation.65 Conversely, ruthenium is expensive, hence the tested catalyst may open an avenue for the formulation of efficient and low-priced methanol oxidation Pt based catalysts.
4. Conclusions
The electro-deposition of the platinum catalyst nanoparticles was performed using the RoDSE technique on Y zeolite and Y zeolite/Vulcan composite supports. The catalyst materials were characterized with electron microscopy, X-ray photoelectron spectrometry, powder X-ray diffraction, thermo-gravimetric analysis, diffuse reflectance Fourier transform infrared spectrometry and carbon dioxide adsorption.TEM showed the presence of Pt nanoparticles and Pt tetrahedral, morphology due to the zeolite framework on nucleation. The XPS data revealed that the majority of the platinum was in its metallic form indicating that the deposition process was very efficient. The TGA, DRIFTS and adsorption data demonstrated that the zeolite included in the Pt/Y/V catalyst conserved their structure after all the treatments suffered by this materials, then exhibiting a microporous channel system containing acid bridging hydroxyls on their surface.
The voltammograms corresponding to Pt/Y/V, 40 wt% Pt/Vulcan XC-72R, and Pt black catalysts exhibited a similar behavior. In basic media, the Pt/Y/V catalyst exhibited current density values significantly higher for methanol oxidation than that of the 40 wt% Pt/Vulcan XC-72R, and Pt black catalysts. Since, it is acknowledged that the electro-catalytic activity for the tested process is determined by the accumulation rate of adsorbed CO, and the H–Y zeolite is an excellent adsorbent, consequently, is rational to assume that the mechanism producing the high activity of the Pt/V/Y catalyst is CO2 adsorption by the zeolite phase included in the catalyst formulation.
The platinum catalyst is normally alloyed with Ru, Sn, W, Mo, and other metals to produce bifunctional sites in the catalyst surface and reduce the CO poisoning. Platinum–ruthenium alloys nanoparticles are considered the most active electro-catalysts for methanol oxidation; however ruthenium is very expensive in comparison to a Na–Y zeolite. Consequently, the inclusion of a zeolite in the electro-catalyst opens an avenue for the design of catalysts that may reduce CO poisoning.
Acknowledgements
This work was partially funded by NASA-URC Grant Number NNX10AQ17A. The authors R.P. and R.R.M. (University of Turabo) acknowledge the financial support provided by the US Department of Energy through the Massey Chair project and the National Science Foundation (CHE-0959334). We thank Ms Frances Lugo, Mr Ian Gutierrez Molina, Mr Carlos Muñiz and the Puerto Rico Energy Center. NSF for its support (award 0701525) to the Nanoscopy Facility, an electron microscopy facility at UPR.References
- P. Jena, J. Phys. Chem. Lett., 2011, 2, 206–211 CrossRef CAS.
- H. S. Wang and H. D. Abruna, in Fuel Cells and Hydrogen Storage, ed. A. Bocarsly and D. M. P. Mingos, 2011, vol. 141, pp. 33–83 Search PubMed.
- D. Chu and R. Z. Jiang, Solid State Ionics, 2002, 148, 591–599 CrossRef CAS.
- C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183–4206 CrossRef CAS PubMed.
- E. H. Yu, U. Krewer and K. Scott, Energies, 2010, 3, 1499–1528 CrossRef CAS PubMed.
- R. Z. Jiang and D. R. Chu, J. Electrochem. Soc., 2004, 151, A69–A76 CrossRef CAS PubMed.
- E. Antolini, Appl. Catal., B, 2009, 88, 1–24 CrossRef CAS PubMed.
- A. D. Moore, S. M. Holmes and E. P. L. Roberts, RSC Adv., 2012, 2, 1669–1674 RSC.
- S. C. Thomas, X. M. Ren, S. Gottesfeld and P. Zelenay, Electrochim. Acta, 2002, 47, 3741–3748 CrossRef CAS.
- E. Antolini, Mater. Chem. Phys., 2003, 78, 563–573 CrossRef CAS.
- S. Wasmus and A. Kuver, J. Electroanal. Chem., 1999, 461, 14–31 CrossRef CAS.
- X. L. Sui, Z. B. Wang, M. Yang, L. Huo, D. M. Gu and G. P. Yin, J. Power Sources, 2014, 255, 43–51 CrossRef CAS PubMed.
- D. Santiago, G. G. Rodriguez-Calero, H. Rivera, D. A. Tryk, M. A. Scibioh and C. R. Cabrera, J. Electrochem. Soc., 2010, 157, F189–F195 CrossRef CAS PubMed.
- D. Santiago, G. G. Rodriguez-Calero, A. Palkar, D. Barraza-Jimenez, D. H. Galvan, G. Casillas, A. Mayoral, M. Jose-Yacaman, L. Echegoyen and C. R. Cabrera, Langmuir, 2012, 28, 17202–17210 CrossRef CAS PubMed.
- W. Z. Li, C. H. Liang, J. S. Qiu, W. J. Zhou, H. M. Han, Z. B. Wei, G. Q. Sun and Q. Xin, Carbon, 2002, 40, 791–794 CrossRef CAS.
- C. M. Chang, H. Y. Li, J. Y. Lai and Y. L. Liu, RSC Adv., 2013, 3, 12895–12904 RSC.
- G. S. Chai, S. B. Yoon, J. S. Yu, J. H. Choi and Y. E. Sung, J. Phys. Chem. B, 2004, 108, 7074–7079 CrossRef CAS.
- L. La-Torre-Riveros, R. Guzmán-Blas, A. E. Mendez-Torres, M. Prelas, D. A. Tryk and C. R. Cabrera, ACS Appl. Mater. Interfaces, 2012, 4, 1134–1147 CAS.
- K. V. Kordesch and G. R. Simader, Chem. Rev., 1995, 95, 191–207 CrossRef CAS.
- T. M. Salama, I. O. Ali and H. A. Gumaa, Microporous Mesoporous Mater., 2008, 113, 90–98 CrossRef CAS PubMed.
- Z. Tang, J. Monroe, J. Dong, T. Nenoff and D. Weinkauf, Ind. Eng. Chem. Res., 2009, 48, 2728–2733 CrossRef CAS.
- P. Kaminski, I. Sobczak, P. Decyk, M. Ziolek, W. J. Roth, B. Campo and M. Daturi, J. Phys. Chem. C, 2013, 117, 2147–2159 CAS.
- D. R. Rolison and C. A. Bessel, Acc. Chem. Res., 2000, 33, 737–744 CrossRef CAS PubMed.
- D. R. Rolison, Chem. Rev., 1990, 90, 867–878 CrossRef CAS.
- D. R. Rolison, Advanced Zeolite Science and Applications, 1994, vol. 85, pp. 543–586 Search PubMed.
- M. Hernandez-Vélez and R. Roque-Malherbe, J. Mater. Sci. Lett., 1995, 14, 1112–1114 CrossRef.
- T. M. Mudrinić, Z. D. Mojović, A. Z. Ivanović-Šašić, N. S. Vukelić, Ž. D. Čupić and D. M. Jovanović, Russ. J. Phys. Chem., 2013, 87, 2127–2133 CrossRef.
- O. Vigil, J. Fundora, H. Villavicencio, M. Hernandez-Velez and R. Roque-Malherbe, J. Mater. Sci. Lett., 1992, 11, 1725–1727 CrossRef CAS.
- R. Roque-Malherbe, Adsorption and Diffusion in Nanoporous Materials, CRC Press, Boca Raton, FL, USA, 2007 Search PubMed.
- D. W. Breck, Zeolite Molecular Sieves, Wiley-Interscience, J. Wiley & Sons, New York, 1974 Search PubMed.
- R. Roque-Malherbe, Physical Chemistry of Materials Energy and Environmental Applications, CRC Press, Boca Raton, FL, USA, 2009 Search PubMed.
- G. R. Li, H. Xu, X. F. Lu, J. X. Feng, Y. X. Tong and C. Y. Su, Nanoscale, 2013, 5, 4056–4069 RSC.
- M. C. Silaghi, C. Chizallet and P. Raybaud, Microporous Mesoporous Mater., 2014, 191, 82–96 CrossRef CAS PubMed.
- T. C. Gruber, T. W. Zerda and M. Gerspacher, Carbon, 1993, 31, 1209–1210 CrossRef CAS.
- G. Selvarani, S. V. Selvaganesh, S. Krishnamurthy, G. V. M. Kiruthika, P. Sridhar, S. Pitchumani and A. K. Shukla, J. Phys. Chem. C, 2009, 113, 7461–7468 CAS.
- R. L. Barbosa, V. Papaefthimiou, Y. T. Law, D. Teschner, M. Havecker, A. Knop-Gericke, R. Zapf, G. Kolb, R. Schlogl and S. Zafeiratos, J. Phys. Chem. C, 2013, 117, 6143–6150 CAS.
- D. C. Higgins, D. Meza and Z. W. Chen, J. Phys. Chem. C, 2010, 114, 21982–21988 CAS.
- R. Roque-Malherbe, O. N. C. Uwakweh, C. Lozano, R. Polanco, A. Hernandez-Maldonado, P. Fierro, F. Lugo and J. N. Primera-Pedrozo, J. Phys. Chem. C, 2011, 115, 15555–15569 CAS.
- J. W. Arblaster, Platinum Met. Rev., 1997, 41, 12–21 CAS.
- X. W. Yu and S. Y. Ye, J. Power Sources, 2007, 172, 133–144 CrossRef CAS PubMed.
- M. M. Treacy and J. B. Higgins, Collection of Simulated XRD Powder Patterns for Zeolites, Elsevier, Amsterdam, 2001 Search PubMed.
- A. Dyer, Thermochim. Acta, 1987, 110, 521–526 CrossRef CAS.
- W. Martinez, T. T. Thompson and M. A. Smit, Int. J. Electrochem. Sci., 2010, 5, 931–943 Search PubMed.
- H. Knozinger and S. Huber, J. Chem. Soc., Faraday Trans., 1998, 94, 2047–2059 RSC.
- J. Matthey, http://www.chemicals.matthey.com/userfiles/files/Pt(1).pdf, 2009.
- A. Zecchina and C. O. Arean, Chem. Soc. Rev., 1996, 25, 187–197 RSC.
- J. A. Lercher, C. Grundling and G. EderMirth, Catal. Today, 1996, 27, 353–376 CrossRef CAS.
- R. Roque-Malherbe, R. Polanco-Estrella and F. Marquez-Linares, J. Phys. Chem. C, 2010, 114, 17773–17787 CAS.
- B. P. Bering, M. M. Dubinin and V. V. Serpinsky, J. Colloid Interface Sci., 1972, 38, 185–194 CrossRef CAS.
- S. Trasatti and O. A. Petrii, Pure Appl. Chem., 1991, 63, 711–734 CrossRef CAS.
- C. Korzeniewski, V. Climent and J. M. Feliu, Electroanalytical Chemistry: A Series of Advances, 2012, vol. 24, pp. 75–169 Search PubMed.
- N. M. Markovic and P. N. Ross, Surf. Sci. Rep., 2002, 45, 121–229 CrossRef.
- C. M. Sanchez-Sanchez, J. Solla-Gullon, F. J. Vidal-Iglesias, A. Aldaz, V. Montiel and E. Herrero, J. Am. Chem. Soc., 2010, 132, 5622–5624 CrossRef CAS PubMed.
- R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343–1348 CrossRef CAS.
- B. Lim, M. J. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. M. Lu, Y. M. Zhu and Y. N. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- J. Solla-Gullon, F. J. Vidal-Iglesias, A. Lopez-Cudero, E. Garnier, J. M. Feliu and A. Aldaz, Phys. Chem. Chem. Phys., 2008, 10, 3689–3698 RSC.
- J. Kim, C. Jung, C. K. Rhee and T. H. Lim, Langmuir, 2007, 23, 10831–10836 CrossRef CAS PubMed.
- E. Herrero, K. Franaszczuk and A. Wieckowski, J. Phys. Chem., 1994, 98, 5074–5083 CrossRef CAS.
- L. Li and Y. C. Xing, Energies, 2009, 2, 789–804 CrossRef CAS PubMed.
- N. M. Markovic, C. A. Lucas, B. N. Grgur and P. N. Ross, J. Phys. Chem. B, 1999, 103, 9616–9623 CrossRef CAS.
- C. Muñiz-López, J. Duconge and R. Roque-Malherbe, J. Colloid Interface Sci., 2009, 329, 11–16 CrossRef PubMed.
- R. F. Lobo, Handbook of Zeolite Science and Technology, ed. S. M. Auerbach, K. A. Carrado and P. K. Dutta, Marcell Dekker Inc., New York, 2003, pp. 65–69 Search PubMed.
- R. Roque-Malherbe and F. Diaz-Castro, J. Mol. Catal. A: Chem., 2008, 280, 194–202 CrossRef CAS PubMed.
- H. S. Wang, L. R. Alden, F. J. DiSalvo and H. D. Abruna, Langmuir, 2009, 25, 7725–7735 CrossRef CAS PubMed.
- S. L. Gojkovic, T. R. Vidakovic and D. R. Durovic, Electrochim. Acta, 2003, 48, 3607–3614 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |