
Co2+-loaded periodic mesoporous aluminum phosphonates for efficient modified Fenton catalysis†
Yun-Pei Zhu a,
Tie-Zhen Renb and
Zhong-Yong Yuan*a
a,
Tie-Zhen Renb and
Zhong-Yong Yuan*a
aKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: zyyuan@nankai.edu.cn
bSchool of Chemical Engineering and Technology, Hebei University of Technology, Tianjin 300130, China
First published on 19th December 2014
Abstract
Periodic mesoporous aluminum phosphonate (PMAP) materials with homogeneously integrated organophosphonate bridging groups inside the hybrid framework were synthesized by an autoclaving process using ethylene diamine tetra(methylene phosphonic acid) as the coupling molecule, with the assistance of the cationic surfactant cetyltrimethylammonium bromide. The prepared aluminum phosphonates possessed a high specific surface area of 511 m2 g−1 and a typical hexagonal mesophase, thus guaranteeing the considerable uptake capacity for loading Co2+ ions through coordination interaction. The monolayered adsorption behavior of Co2+ was confirmed, and the Co2+-loaded PMAP could be further utilized as a heterogeneous catalyst for oxidizing decomposition of phenol in the presence of peroxymonosulfate, with favorable kinetic and thermodynamic characteristics. It is suggested that the functionalities of metal phosphonate organic–inorganic hybrids could be rationally designed by judiciously selecting precursors and post-modification, making them potentially applicable in environmental remediation and catalysis.
1. Introduction
Environmentally-friendly catalysis is among the most significant applications within the area of nanoscience. Efficient destruction of organic contaminants in waste water involving advanced oxidation processes (AOPs) has received great scientific and technological interests in environmental applications during last decades.1–4 AOPs involve the generation of highly active oxidizing species that attack and decompose organic substances, making the oxidizing processes superior over other techniques based on physical processes including adsorption and flocculation. Classical homogeneous Fenton reagent consisting of Fe2+ and hydrogen peroxide has been testified to be of valuable degradation efficiency and general applicability, though the requirement of acidic conditions to avoid ferrous and ferric ion hydrolysis and the removal of sludge containing iron ions make this method unamiable and uneconomical.5,6 Driven by the need of overcoming the drawbacks of Fenton reagent and seeking for another alternative system that can introduce oxidants outperforming the hydroxyl radicals, a variety of modified Fenton systems have been proposed, such as the manganese-, copper-, nickel-based catalysts involving H2O2;7,8 nevertheless, their further application is blocked by the insufficient catalytic performance. Remarkably, among various transition and noble metal ions, Co2+ exhibits considerable capability for the activation of peroxymonosulfate (PMS) to produce sulfate radicals, due to the suitable redox potential.5,9–11 The more powerful oxidizing capability of sulfate radicals than hydroxyl counterparts at neutral pH and their similarity at acidic pH could be related to the abilities of their redox partners as leaving groups, wherein the bisulfate and sulfate ions are for the active sulfate radical and the water molecule for the hydroxyl radical.12 However, it should be noted that the toxicity associated with cobalt ions during homogeneous reaction leads to further health concern and pollution. On the other hand, recovery and reuse of the catalysts after catalytic reactions are important factors for sustainable process management, while homogeneous Fenton systems are prohibited to a wide application range from this point of view. Moreover, the alleviation of damage from organic contaminants and cobalt ions simultaneously through a facile route remains a challenge.Heterogenization of the homogeneous catalysts provides a myriad of advantages including ease separation and catalyst reusability. For cobalt-based catalytic systems, several heterogeneous catalysts have been investigated such as Co3O4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13,14 and CoFeO4 nanoparticles,15 exhibiting obvious Co2+ leaching. Further attempts were made to immobilize the active cobalt species on various supporters, for instance, carbon nanotubes,16 graphene oxide,17 activated carbon,18 magnesium oxide,19 and titanium oxide.20 But the corresponding energy-intensive preparation technologies, high cost of the supporting materials and the insufficient catalytic activity subjected to the poor porosity make them find difficulties in the practical applications.
13,14 and CoFeO4 nanoparticles,15 exhibiting obvious Co2+ leaching. Further attempts were made to immobilize the active cobalt species on various supporters, for instance, carbon nanotubes,16 graphene oxide,17 activated carbon,18 magnesium oxide,19 and titanium oxide.20 But the corresponding energy-intensive preparation technologies, high cost of the supporting materials and the insufficient catalytic activity subjected to the poor porosity make them find difficulties in the practical applications.
Well-defined mesoporosity accompanied by high specific surface area and large pore volume not only can facilitate interaction between active sites and guest molecules at the pore surface and throughout the bulk of the material, but also promote mass transfer of intermediates, resulting in tremendous potential in the fields of catalysis, adsorption and biotechnology.21,22 Chemically designed metal phosphonate hybrid mesoporous materials have been proven to be multifunctional due to the combined physicochemical superiorities from inorganic and organic moieties, thereby presenting fantastic surface chemistry for meaningful practicability, and intentional introduction of well-structured mesoporosity by using surfactants could further improve the apparent performances.23–25 As one of the classical applications, heavy metal ion adsorption for metal phosphonates has attracted wide attention on the basis of that the organic functionalities in the hybrid network typically serve to form complexes with heavy metal ions through acid–base reactions.21,26 The adsorption behavior of metal ions for the phosphonate-based hybrid adsorbents was found to follow Langmuir-type monolayer model due to the uniform bridging between organic linkages and inorganic units.26,27 It is reasonable to envision that the toughly immobilized metal ions can act as catalytically active sites for specular reactions, thus achieving the heterogenization of homogeneous catalysts, which is beneficial to the recyclability and reusability of the homogeneous catalytic species. However, there are still scarce reports that realize such an objective.
Herein, periodic mesoporous aluminum phosphonates with high specific surface area were synthesized with the assistance of cetyltrimethylammonium bromide (CTAB) through an autoclaving process, using ethylene diamine tetra(methylene phosphonic acid) (EDTMP) as the organic bridging groups. The homogeneously allocated ethylene diamine functional groups on the mesoporous channels demonstrated high capability to coordinate cobalt ions, realizing the heterogenization of homogeneous active Co2+ ions on the hybrid support. A step beyond would be promising in oxidizing decomposition of organic pollutants in the presence of PMS.
2. Experimental
2.1 Chemicals
Aluminium trichloride (AlCl3), cetyltrimethylammonium bromide (CTAB), phenol, and Co(NO3)2·6H2O were obtained from Tianjin Guangfu Chemical Co. EDTMP was received from Shandong Taihe Chemical Co., Ltd. Potassium peroxymonosulfate was received from Tianjin Shengmiao Chemical Co. All chemicals were used as received without further purification.2.2 Synthesis of periodic mesoporous aluminum phosphonate
In a typical run, 1.6 mmol of EDTMP and 2.0 mmol of CTAB were dissolved in the mixed solution of 20 mL of H2O and 5 mL of ethanol, followed by vigorous stirring for 2 h. Thereafter, AlCl3 was added into the mixed solution very slowly (P/Al molar ratio: 5/4), and the pH value of the mixture was kept around 9.0 through the entire process by ammonia and HCl. Then the homogeneous mixture was transferred into a 50 mL-sized Teflon-lined autoclave and aged statically at 110 °C under autogenous pressure for 36 h. After cooled naturally to room temperature, the resultant mixtures were filtered and dried. The removal of the surfactant was accomplished by extracting the as-synthesized material in ethanol (200 mL) with concentrated HCl (4.2 g) for 24 h, and the final sample was marked as PMAP. Meanwhile, aluminum phosphonate counterpart with disordered mesoporosity was synthesized through the similar way while in the absence of surfactant, and labeled as MAP.2.3 Characterization
X-ray diffraction (XRD) patterns were recorded on a Bruker D8 Focus Diffractometer with Cu-Kα radiation (λ = 0.15418 nm) operated at 40 kV and 40 mA. Scanning electron microscopy (SEM) was carried out on a Jeol JSF-7500L microscope at 5 kV. Transmission electron microscopy (TEM) was carried out on a Jeol JEM 2100F microscope at 200 kV. All samples subjected to TEM measurements were ultrasonically dispersed in ethanol and dropcast onto copper grids covered with a carbon film. N2 adsorption–desorption isotherms were measured on a Quantachrome NOVA 2000e sorption analyzer at liquid nitrogen temperature (77 K). Prior to measurement, the samples were degassed at 120 °C overnight. Surface areas were calculated by the multi-point Brunauer–Emmett–Teller (BET) method. Fourier transform infrared (FT-IR) spectra were measured on a Bruker VECTOR 22 spectrometer with KBr pellet technique, and the ranges of spectrograms were 4000 to 400 cm−1. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Kratos Axis Ultra DLD (delay line detector) spectrometer equipped with a monochromatic Al-Kα X-ray source (1486.6 eV). All XPS spectra were recorded using an aperture slot of 300 × 700 microns, survey spectra were recorded with a pass energy of 160 eV and high resolution spectra with a pass energy of 40 eV. Thermogravimetry analysis (TGA) was performed using a TA SDT Q600 instrument at a heating rate of 5 °C min−1 using α-Al2O3 as the reference. The chemical compositions of Al and P were analyzed by inductively coupled plasma (ICP) emission spectroscopy on a Thermo Jarrell-Ash ICP-9000 (N+M) spectrometer, and C, N, and H were analyzed on a Vario-EL elemental analyzer.2.4 Metal ion adsorption
The ability of PMAP for Co2+ adsorption was tested as follows: 2 mg mL−1 of the samples were added into Co(NO3)2 buffered solution (pH = 6.0, phthalate) with different concentrations of 0.2 to 1.8 × 10−4 mol L−1. After stirring for 8 h to access the adsorption–desorption balance, the mixtures were filtered and the residual metal ion concentrations in the filtrates were analyzed by graphite furnace atomic absorption spectroscopy (AAS). The adsorption capacities of the adsorbent were then determined from the concentration difference measured between the filtrates and the initial metal ion solutions. The maximal adsorption capacity was calculated by the Langmuir model using the equation ns = Knmc/(1 + Kc), where K is the Langmuir constant, c is the Co2+ concentration, nm is the monolayer adsorption capacity, and ns is the amount of Co2+ adsorbed on the adsorbent. The distribution coefficient (Kd) was determined using the equation Kd = (ci − cf)V/(cfmads), where ci is the initial metal ion concentration, cf is the ion concentration after adsorption, V is the volume of the solution (in mL), and mads is the amount of adsorbent (in g).2.5 Preparation of Co2+@PMAP
The determined monolayer adsorption capacity was considered as the standard for loading Co2+ ions. Typically, 1 g of the PMAP sample was added in 10 mL of Co(NO3)2 buffered solution (pH = 6.0, 1.0 × 10−4 mol L−1) under gentle magnetic stirring for 8 h to achieve the Co2+ adsorption saturation. The mixture was filtered, and the resultant Co2+-loaded PMAP material was washed with water and ethanol alternatively to remove excess Co2+ ions, and dried at 80 °C under vacuum conditions for 12 h. The ultimate material was denoted as Co2+@PMAP.2.6 Catalytic oxidation tests
The catalytic activity of the prepared cobalt phosphonate materials was evaluated by the degradation of phenol with the assistance of PMS. Typically, 20 mg of Co2+@PMAP was added to 50 mL of 10 ppm phenol solution, and the degradation of phenol was initiated using PMS oxidizing agent at a concentration of 6 mmol L−1. Aliquots of reaction solutions were sampled at designated time intervals during the reaction. The absorbance of reaction solutions was measured using a SP-722 spectrometer at λmax = 504 nm. To evaluate the stability of the catalysts, the sample after one trial was collected through centrifugation, washed by water and ethanol alternatively, and dried for the subsequent cycle test.3. Results and discussion
3.1 Material synthesis and characterization
Periodic mesoporous aluminum phosphonate material was prepared through a cationic surfactant-assisted sol–gel strategy, wherein AlCl3 was added slowly into the mixed solution of CTAB and EDTMP. The pH value of the reaction system should be kept at around 9.0, which was found to be a crucial factor to obtain well-developed mesostructures. The XRD patterns of the aluminum phosphonates prepared in the presence of CTAB are shown in Fig. 1. The low-angle XRD pattern of PMAP demonstrates a typical hexagonal mesophase with a major diffraction situated at 2θ = 2.08° assigned to (100) reflection, accompanied with two well-resolved peaks at 2θ = 3.65° and 4.16° corresponding to (110) and (200) reflections, respectively. The unit cell parameter (a) was calculated to be 4.9 nm. The periodic mesostructure was further confirmed by TEM and N2 sorption analysis. The hexagonal arrangement of mesopores with pore size of about 3.1 nm can be clearly observed in Fig. 2a, which is similar to the previously reported aluminum phosphonates prepared with the assistance of CTAB.28 Moreover, typical one-dimensional channels can be seen from the side view of PMAP (Fig. 2b). The N2 sorption isotherm of PMAP is of type IV without visible hysteresis loop (Fig. 3), suggesting reversible capillary condensation–evaporation in mesopores, which is typical for some OOINs (ordered organic–inorganic nanocomposites) with accessible mesostructures.29 The resulting pore size distribution curve determined by Non-Local Density Functional Theory (NLDFT) model presents a narrow peak around 3.1 nm, coinciding with the TEM observation. The specific surface area and pore volume are calculated to be 511 m2 g−1 and 0.401 cm3 g−1, respectively. The formation mechanism of the prepared ordered mesoporous aluminum phosphonate hybrid material with the assistance of cationic surfactant CTAB under weakly alkaline condition is proposed that the positive charge-associated CTA+ micelles and the anionic aluminum phosphonate species were assembled together under the drive of electrostatic and van der Waals interactions, similar to the S+I− ionic mechanism.30,31 This is first example involving the successful ordered mesoporous metal phosphonates under basic conditions to the best of our knowledge. | ||
| Fig. 1 Low and wide angle XRD patterns of the synthesized PMAP and Co2+@PMAP materials. | ||
 | ||
| Fig. 2 TEM images of the PMAP sample. | ||
 | ||
| Fig. 3 N2 adsorption–desorption isotherms (a) of PMAP and Co2+@PMAP, and the corresponding pore size distribution curves (b) determined by the NLDFT model. | ||
The skeleton structure and surface chemistry of the mesoporous hybrid were investigated by FT-IR and XPS. The FT-IR spectra of the PMAP sample and coupling molecule EDTMP are shown in Fig. 4. The strong broad band at 3400 cm−1 and the sharp band at 1653 cm−1 correspond to the surface-adsorbed water and hydroxyl groups. The strong band at 1053 cm−1 is due to phosphonate P–O–Al stretching vibrations. The overlapped bands at 1466 and 1432 cm−1 are assigned as the C–H bending in –CH2– groups and the P–C stretching vibrations, respectively, and the small band at 1317 cm−1 could be attributed to C–N stretching.32 The signal presented at 740–745 cm−1 suggests the presence of P–O–P bending modes. In addition, weak bands around 2800–3000 cm−1 are assigned to the C–H stretching modes. The band at 927 cm−1 in the spectrum of EDTMP, assigned to P–OH stretching vibrations, is not observed in PMAP, which implies the extensive condensation and coordination of the phosphoryl oxygen with the aluminum atom, leading to mainly bidentate phosphonate units.
 | ||
| Fig. 4 FT-IR spectra of EDTMP and PMAP. | ||
High-resolution XPS spectra were also recorded on the surface of the PMAP sample for the investigation of chemical state and surface stoichiometry (Fig. 5). The surface atomic composition of the materials was calculated to be 7.48% Al, 9.21% P, 15.62% C, 61.75% O and 4.94% N for PMAP. The surface P/Al ratio was calculated to be 1.23, approximate to 5/4. The Al 2p line of PMAP sample is composed of a single peak situated at binding energy of 74.5 eV (Fig. S1, ESI†). Compared with the binding energy of pure Al2O3 (75.4 eV for Al 2p),33 the increase of binding energy for the Al 2p in the mesoporous aluminum phosphonate hybrid may be the result from the organophosphonate incorporation in the organic–inorganic network. The P 2p binding energy of PMAP is observed at 131.4 eV (Fig. 5a), which is characteristic of P5+ in phosphonate groups. In the O 1s spectrum (Fig. 5b), the broad signal at 530.3 eV can be attributed to the bridging oxygen in the P–O–Co linkages, and a shoulder around 532.1 eV can be assigned to the surface hydroxyl. The N 1s spectrum (Fig. 5c) shows a main component at 398.6 eV with a shoulder at 401.2 eV, attributable to the bridged N-containing compounds.
 | ||
| Fig. 5 High-resolution XPS spectra of P 2p (a), O 1s (b) and N 1s (c) for PMAP and Co2+@PMAP, and Co 2p (d) for Co2+@PMAP. | ||
The thermal stability of the as-synthesized PMAP hybrid material was investigated through thermal gravimetric analysis (TGA). The TGA curves in Fig. S2† exhibit an initial weight loss of 11.2% from room temperature to 180 °C, which may be assigned to the desorption of the adsorbed and intercalated water. The second weight loss of 10.1% from 180 to 435 °C can be attributable to the decomposition of the surfactant molecules, and the third weight loss of 7.3% from 435 to 650 °C can be associated with the decomposition of the organic functional groups in the hybrid framework. Another small weight loss of 0.5% at beyond 650 °C can be related to the combustion of carbon species. The TGA characterization showed that the hybrid framework could stay stable up to around 435 °C, which is also confirmed by previously reported studies concerning mesoporous aluminium organophosphonates.34,35 The ICP emission spectroscopy was employed to analyze chemical compositions of the resultant solids (13.24% Al, 19.01% P in mass), revealing the P/Al molar ratios approximate to 5:4 in the sample, which was consistent with the ratio of the reagents added, showing the complete condensation of AlCl3 and phosphonic acid. Combined with the conventional elemental analysis of C, H and N (11.04% C, 9.75% H, 4.29% N in mass), the PMAP material could be roughly formulated as Al4(EDTMP)1.25·xH2O.
3.2 Co2+ ion adsorption
The Co2+-loaded PMAP heterogeneous catalyst was prepared by an adsorption strategy. The Co2+ could be immobilized on the hybrid matrix on the basis of coordination effect, and the homogeneously distributed organophosphonate functionalities ensured the high and uniform dispersion on the mesoporous hybrid materials. The Co2+ adsorption behavior for the PMAP sample was thus investigated to determine the saturation amount of Co2+ ions. The PMAP hybrid was treated with a series of Co(NO3)2 aqueous solution with different concentrations. It is found that the adsorption behavior follows Langmuir-type model (Fig. 6a), with the ions being almost quantitatively adsorbed until binding saturation was reached. According to the Langmuir equation, the maximal adsorption capacity was calculated to be 24.8 μmol g−1, which is set as the standard for loading Co2+ ions. As shown in Fig. 6b, The PMAP presents high distribution coefficient (Kd) particularly at low Co2+ concentrations (8591 mL g−1 when treated with 0.2 × 10−4 mol L−1 Co2+ aqueous solution), which decreases with the increase of the initial concentration. This implies the high affinities for the uptake of Co2+ ions at low levels. Noticeably, the resultant adsorption ability outperforms previous reported hierarchically porous titanium phosphonate materials,36 making the present mesoporous aluminum phosphonates promising adsorbents for the heavy metal ion removal.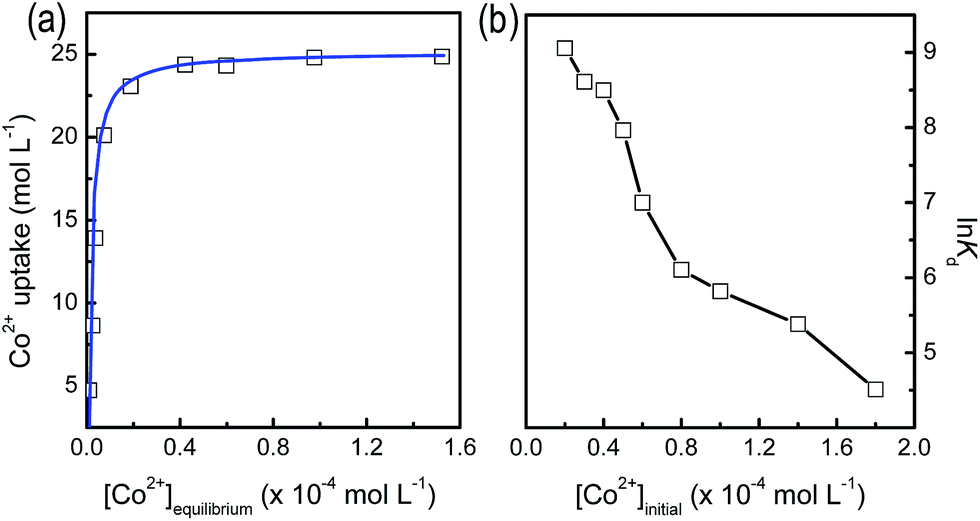 | ||
| Fig. 6 (a) Co2+ ion adsorption isotherm of PMAP, and (b) the corresponding distribution coefficient profile. The solid blue line in (a) is the simulated line using Langmuir equation. | ||
The low-angle XRD pattern of Co2+-loaded PMAP (Co2+@PMAP) (Fig. 1) exhibits three typical peaks related to hexagonal mesophase, i.e., one main peak corresponding to the (100) reflection and two small peaks assigned to (110) and (200) reflections, suggesting the preservation of the long-range ordered mesostructure after incorporating cobalt ions. The wide angle XRD pattern of Co2+@PMAP presents still the amorphous pore walls. No reflection characteristics of cobalt oxides or hydroxies could be observed. The nitrogen adsorption–desorption isotherms of Co2+@PMAP are of type IV with type H2 hysteresis as well, meaning the reservation of well-defined mesopores. The pore size distribution, obtained from the NLDFT model, present a narrow peak at about 3.1 nm, and the surface area of Co2+@PMAP (506 m2 g−1) is a little smaller than that of PMAP (511 m2 g−1), suggesting that the incorporated Co2+ ions would not intensively affect the textural and structural properties of the synthesized hybrid material. The TEM images of Co2+@PMAP (Fig. S3†) also confirmed that the periodic mesoporous structure could be well observed after Co2+ loading.
Meanwhile, UV-Vis absorbance spectrum was conducted to illustrate the interaction between Co2+ ions and the PMAP host material. Fig. 7 indicates that absorbance peak of Co2+@PMAP has a red shift from 203 to 210 nm as compared with that of the pristine PMAP. Notably, a broad absorbance shoulder ranging from 400 to 700 nm can clearly be seen. Coordination between Co2+ and the pore surface of PMAP is regarded as the reason to the variation of UV-Vis absorbance due to the existence of organophosphonate species in the hybrid network,26,37 akin to the coordination effect of the ethylenediamine-functionalized porous silica systems.38,39
 | ||
| Fig. 7 UV-Vis absorbance spectra of PMAP before and after Co2+ ion loading. | ||
In order to further confirm the chemical nature of Co2+ adsorption on PMAP, high-resolution XPS measurements were thus performed. As shown in Fig. S1† and 5, no obvious differences between PMAP and Co2+@PMAP can be observed in the Al 2p, O 1s and P 2p spectrum. However, after being coordinated with Co2+, the two signals of N 1s corresponding to the bridging nitrogen species in the phosphonic linkages shifted to higher binding energy, which can be due to the formation of the coordination bond of N-metal ions where the electron-rich nitrogen atoms shared electrons with Co and thus electron densities of nitrogen atoms were reduced.40 For the high-resolution Co 2p spectrum (Fig. 5d), the binding energies were observed to be 795.8 eV for Co 2p1/2 and 780.6 eV for Co 2p3/2. The spin–orbit splitting value of the 2p peaks was determined to be 14.8 eV, indicating that the immobilized cobalt ions on the mesoporous hybrid wall exist as divalent species.41 Moreover, relatively intense satellite peaks at 785.7 and 800.8 eV reveal the presence of divalent Co since mixed-valence exhibits a weak satellite structure.42,43 In fact, the claw tetraphosphonic acid could react with metal ions. The complete chelation between phosphonic acids with aluminum was confirmed by the FT-IR and XPS characterization. Only scare free P–OH ligands were left after the aluminum phosphonate hybrid framework was generated, and thus Co2+ adsorption was mainly due to coordination of the N atoms (Fig. 8), which was previously reported for the ethylene-diamine integrated PMOs-based adsorbents.36,37 The surface atomic composition of Co2+@PMAP was calculated to be 7.45% Al, 9.18% P, 15.66% C, 62.47% O, 4.91% N and 0.33% Co. The surface P/Al ratio was calculated to be 5/4, the same as that of PMAP, implying that no change happened to the chemical compositions and the organic groups in the hybrid framework.
 | ||
| Fig. 8 Schematic illustration of the formation process for the Co2+-loaded PMAP material. | ||
To test the reusability of the PMAP adsorbent, the Co2+ ion loaded sample was treated with 1 mol L−1 hydrochloric acid for 8 h to remove the heavy metal ions and then neutralized, following a second round of metal ion adsorption testing. The results for Co2+ adsorption using the regenerated adsorbents are summarized in Fig. S4.† Only a slight deterioration from 98.6 to 94.3% could be observed during the second use, and the sample retained a Co2+ removal efficiency of 92.5% after three cycles. Then the Co2+ uptake capacities decreased gradually with the subsequent successive use, but the hybrid still reserved about 85% of the initial metal ion loading capacity after leaching six times. This signifies the stability of the synthesized periodic mesoporous aluminum phosphonate materials and the retention of their adsorption properties under the relatively strong acid leaching conditions, making them useful as reusable sorbents for multiple metal ion adsorption cycles.
3.3 Catalytic performance
The homogeneous Co2+/PMS systems have been testified to be considerably efficient in decomposing organic contaminants.8–11 The simultaneous incorporation of Co2+ and well-defined mesoporosity in the Co2+@PMAP materials inspired us to investigate the catalytic oxidation capability as a Fenton-like heterogeneous catalyst. Fig. 9a depicts the variation of phenol concentration as a function of time for different systems. To have a better understanding of the reaction kinetics of the phenol degradation catalyzed by the heterogeneous hybrid catalysts, a general pseudo-first-order kinetics for phenol decay could be assumed: ln(C0/C) = kt, where C0/C represents the normalized organic compound concentration and k is the apparent reaction rate constant. As to PMS without a solid catalyst, degradation of phenol is negligible after 60 min, indicating that the PMS itself could not be activated for oxidizing phenol in aqueous solution under ambient conditions. The phenol concentration showed no evident change in the presence of PMAP, which suggested that the consumption of phenol by adsorption process could be ignored. For the PMS/PMAP system, the PMAP material could hardly activate PMS to produce active sulfate species to result in phenol oxidation. Nonetheless, the Co2+@PMAP material could produce significant phenol decomposition with the assistance of PMS, and complete degradation could be achieved within 50 min with the reaction rate constant of 0.0739 min−1. As shown in Fig. S5,† it can be found that the catalytic efficiency increased with the Co2+ loading amount and then reached the maximum after the immobilization ability reached the saturation value. Thus, the mesoporous hybrid materials saturated with Co2+ were chosen in the following tests. | ||
| Fig. 9 (a) Degradation kinetic curves of phenol by the synthesized aluminum phosphonate materials as catalysts under ambient conditions, and (b) the corresponding linear fitting results. (c) Quenching studies for radical identification using different alcohols. (d) Reusability tests of Co2+@PMAP for phenol decomposition. | ||
In general, the activity of catalyst strongly depends on the properties of support. Here, the ordered mesoporous structures with high specific surface area could contribute to the enhancement of mass transfer and supply abundant sites for loading catalytically active Co2+ ions. To confirm this point, the periodic mesoporous structure of Co2+@PMAP was destroyed by a ball-milling treatment, which was denoted as Co2+@d-PMAP, leading to a reduction of surface area to 138 m2 g−1. Although the resultant materials had the same Co2+ loading amount, the oxidizing degradation was decreased to 64.7% after 60 min, accompanied with k = 0.0181 min−1. For the purpose of comparison, mesoporous aluminum phosphonate material with disordered mesostructure (Fig. S6†), labeled as MAP, was synthesized in a similar way to PMAP while in the absence of cationic surfactant. However, the MAP hybrid possessed a relatively lower surface area of 155 m2 g−1 as compared with that of PMAP (511 m2 g−1), and the corresponding Co2+ uptake capability was determined to be 8.13 μmol g−1. The Co2+@MAP could degrade 53.7% phenol (k = 0.0127 min−1), even exhibiting inferior activity in comparison with that of Co2+@d-PMAP. This could be associated with the distinct Co2+ immobilizing amount on these hybrid heterogeneous catalysts.
To identify primary radical species formed during the oxidation decomposition of phenol, quenching experiments were conducted with the addition of specific alcohols. The cobalt-mediated activation of PMS is known to generate three different radicals, hydroxyl, sulfate and PMS.5,44 It is generally considered that alcohols containing alpha hydrogen, such as ethanol, react at high and comparable rates with hydroxyl and sulfate radicals, and alcohols without alpha hydrogen, including tert-butyl alcohol, exhibit high efficiency in quenching hydroxyl radicals but inefficiency in consuming sulfate radicals. However, PMS radicals are relatively inert towards alcohols. According to these properties, the real active radical during the catalytic process can be easily identified through the addition of particular alcohols. Fig. 9c demonstrates that the addition of ethanol into the reaction solution results in a remarkable decrease of efficiency to 12.7% after 60 min, indicating that PMS radicals could be excluded from being the primary species. Furthermore, the use of tert-butyl alcohol can differentiate the roles of hydroxyl and sulfate radicals. However, the degradation of phenol is slightly affected in the presence of tert-butyl alcohol because only about 9% reduction in degradation efficiency is observed after 50 min. This implies that sulfate radicals are the major oxidizing species formed that are indispensable for the transformation of the substrate. The schematic diagram of the oxidation decomposition process is presented in Fig. 10.
 | ||
| Fig. 10 Scheme of the phenol decomposition process on the pore surface of Co2+@PMAP in the presence of PMS. | ||
As the stability of catalytic activity is another parameter to evaluate a heterogeneous catalyst, the catalytic activity of Co2+@PMAP for phenol oxidizing degradation was tested as a reaction time (Fig. 9d), where the reactions were carried out under ambient conditions. It is found that the reaction rate of phenol decomposition decreased from 0.0739 to 0.0476 min−1 after three times cycling. The analysis of the solution showed that the reaction solution contained 3.2 ppm cobalt ion after the first round and 2.7 ppm cobalt ion leaked in the second run, implying that the loaded cobalt ion was partially leached out after the catalytic reaction. Interestingly, after the Co2+ was reloaded onto the repetitiously used Co2+@PMAP, the reloaded Co2+@PMAP almost recovered its catalytic activity. It has been proved that divalent cobalt ion was the best catalyst for activation of PMS because of the suitable redox potential.5,10 The mechanism of cobalt-based modified Fenton reactions is considered to involve a single electron transfer process, the oxidation of Co(II) with PMS and the formation of sulfate radicals as well as the reduction of Co(III) and the generation of PMS radicals. Quenching experiments testified that the latter transient species were very weak compared to the sulfate ones to induce any degradation of organic pollutants. The active sulfate radicals showed high oxidization ability towards decomposing organics into CO2, H2O and other inorganic products. Protonized hydrogen could be generated during the catalysis process, which might cause the Co2+ leaching and thus result in the catalytic activity deterioration. In present work, periodic mesoporous aluminum phosphonate hybrid materials with hexagonally aligned mesopores and high specific surface area demonstrate considerable capacity for cobalt ion removal. And the Co2+ functionalization endows the mesoporous aluminum phosphonate materials with valuable catalytic activity in the area of environmental remediation.
Reaction temperature is a key factor that influences catalyst activity and phenol removal. Catalytic oxidation reactions were conducted at various temperatures of 25 to 45 °C to investigate the reaction thermodynamics, as shown in Fig. 11. The degradation rate would increase significantly with the increase of the reaction temperature. According to the first-order kinetic model, the reaction rate constants at different temperatures can be obtained and the relationship is found to follow the Arrhenius equation: lnk = lnA + Ea/RT, where Ea is the apparent activation energy, R is molar gas constant and T is reaction temperature. The activation energy of Co2+@PMAP for the heterogeneous phenol disintegration is calculated to be 51.8 kJ mol−1, which is lower than most of previously reported supported cobalt-based catalysts in activation of PMS for organic contaminants degradation (Table 1).18,45–51
 | ||
| Fig. 11 Effect of reaction temperature on phenol decomposition on the Co2+@PMAP sample. | ||
| Material | Apparent rate constant, k/min−1 | Activation energy, Ea/kJ mol−1 | Ref. |
|---|---|---|---|
| Co2+@PMAP | 0.0739 | 51.8 | This work |
| Co/activated carbons | 0.0827 | 59.7 | 18 |
| Co/carbon aerogel | 0.0289 | 62.9 | 45 |
| Co/SiO2 | — | 61.7–75.5 | 46 |
| Co/ZSM-5 | 0.000125 | 69.7 | 47 |
| Co3O4/red mud | 0.0428 | 66.3 | 48 |
| Co3O4/fly ash | 0.00513 | 47.0 | 48 |
| Co/SBA-15 | — | 67.4–81.4 | 49 |
| Co3O4/coal ash | 0.0111 | 56.5 | 50 |
| Co/Fe2O3/CS | 0.0683 | 49.5 | 51 |
4. Conclusions
Periodic mesoporous aluminum phosphonate hybrid material of hexagonal mesostructure for cobalt ions was prepared through a surfactant-mediated strategy using the nitrogen-contained tetra-phosphonic acid. The homogeneously integrated phosphonate bridging groups in the hybrid framework and large surface area realized the high capability for Co2+ ion adsorption. The resultant Co2+-loaded ordered mesoporous hybrid substrates could be further employed as efficient heterogeneous catalysts for oxidizing degradation of organic contaminants with the assistance of peroxymonosulfate. The mesoporous aluminum phosphonates show tremendous potential in the area of sustainable environment development. Furthermore, various metal ions can be immobilized onto the pore wall, and a step beyond including reduction and oxidation makes the organic–inorganic metal phosphonate materials alternative in organic synthesis and photoelectrochemistry.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21421001 and 21073099), the Program for Innovative Research Team in University (IRT13022), the 111 project (B12015), and the Ph.D. Candidate Research Innovation Fund of Nankai University.Notes and references
- E. Neyens and J. Baeyens, J. Hazard. Mater., 2003, 98, 33–50 CrossRef CAS.
- X. J. Long, Z. Yang, H. Wang, M. Chen, K. Y. Peng, Q. F. Zeng and A. H. Xu, Ind. Eng. Chem. Res., 2012, 51, 11998–12003 CrossRef CAS.
- S. H. Tian, Y. T. Yu, D. S. Chen, X. Chen and Y. Xiong, Chem. Eng. J., 2011, 169, 31–37 CrossRef CAS PubMed.
- J. V. Coelho, M. S. Guedes, R. G. Prado, J. Tronto, J. D. Ardissonc, M. C. Pereirad and L. C. A. Oliveir, Appl. Catal., B, 2014, 144, 792–799 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS.
- P. E. F. Oliveira, L. D. Oliveira, J. D. Ardisson and R. M. Lago, J. Hazard. Mater., 2011, 194, 393–398 CrossRef CAS PubMed.
- C. Yu, G. Li, L. Wei, Q. Fan, Q. Shu and J. C. Yu, Catal. Today, 2014, 224, 154–162 CrossRef CAS PubMed.
- A. H. Gemeay, I. A. Mansour, R. G. El-Sharkawy and A. B. Zaki, J. Mol. Catal. A: Chem., 2003, 193, 109–120 CrossRef CAS.
- A. Xu, X. Li, S. Ye, G. Yinc and Q. Zeng, Appl. Catal., B, 2011, 102, 37–43 CrossRef CAS PubMed.
- J. Madhavan, P. Maruthamuthu, S. Murugesan and M. Ashokkumar, Appl. Catal., A, 2009, 368, 35–39 CrossRef CAS PubMed.
- T. Warang, N. Patel, R. Fernandes, N. Bazzanella and A. Miotello, Appl. Catal., B, 2013, 132–133, 204–211 CrossRef CAS PubMed.
- S. Steenken, in Free Radicals: Chemistry, Pathology and Medicine, ed. C. Rice-Evans and T. Dormandy, Richelieu Press, London, 1988, pp. 51–73 Search PubMed.
- G. P. Anipsitakis, E. Stathatos and D. D. Dionysiou, J. Phys. Chem. B, 2005, 109, 13052–13055 CrossRef CAS PubMed.
- X. Y. Chen, J. W. Chen, X. L. Qiao, D. G. Wang and X. Y. Cai, Appl. Catal., B, 2008, 80, 116–121 CrossRef CAS PubMed.
- J. Deng, Y. S. Shao, N. Y. Gao, C. Q. Tan, S. Q. Zhou and X. H. Hua, J. Hazard. Mater., 2013, 262, 836–844 CrossRef CAS PubMed.
- A. Rodriguez, G. Ovejero, J. L. Sotelo, M. Mestanza and J. Garci, Ind. Eng. Chem. Res., 2010, 49, 498–505 CrossRef CAS.
- P. H. Shi, R. J. Su, S. B. Zhu, M. C. Zhu, D. X. Li and S. H. Xu, J. Hazard. Mater., 2012, 229–230, 331–339 CrossRef CAS PubMed.
- P. R. Shukla, S. B. Wang, H. Q. Sun, H. M. Ang and M. Tade, Appl. Catal., B, 2010, 100, 529–534 CrossRef CAS PubMed.
- W. Zhang, H. L. Tay, S. S. Lim, Y. S. Wang, Z. Y. Zhong and R. Xu, Appl. Catal., B, 2010, 95, 93–99 CrossRef CAS PubMed.
- Q. J. Yang, H. Choi, Y. J. Chen and D. D. Dionysiou, Appl. Catal., B, 2008, 77, 300–307 CrossRef CAS PubMed.
- Y. P. Zhu, T. Z. Ren and Z. Y. Yuan, New J. Chem., 2014, 38, 1905–1922 RSC.
- Y. P. Zhu, T. Y. Ma, Y. L. Liu, T. Z. Ren and Z. Y. Yuan, Inorg. Chem. Front., 2014, 1, 360–383 RSC.
- T. Kimura, J. Nanosci. Nanotechnol., 2013, 13, 2461–2470 CrossRef CAS PubMed.
- T. Y. Ma and Z. Y. Yuan, ChemSusChem, 2011, 4, 1407–1419 CrossRef CAS PubMed.
- J. Wegener, A. Kaltbeitzel, R. Graf, M. Klapper and K. Müllen, ChemSusChem, 2014, 7, 1148–1154 CrossRef CAS PubMed.
- T. Y. Ma, X. Z. Lin and Z. Y. Yuan, J. Mater. Chem., 2010, 20, 7406–7415 RSC.
- T. Y. Ma, X. Z. Lin and Z. Y. Yuan, Chem.–Eur. J., 2010, 16, 8487–8494 CrossRef CAS PubMed.
- J. E. Haskouri, C. Guillem, J. Latorre, A. Beltrán, D. Beltrán and P. Amorós, Chem. Mater., 2004, 16, 4359–4372 CrossRef.
- M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13, 3169–3183 CrossRef CAS.
- B. Z. Tian, X. Y. Liu, C. Z. Yu, J. Fan, L. M. Wang, S. H. Xie, G. D. Stucky and D. Y. Zhao, Nat. Mater., 2003, 2, 159–163 CrossRef CAS PubMed.
- J. E. Haskouri, C. Guillem, J. Latorre, A. Beltrán, D. Beltrán and P. Amorós, Eur. J. Inorg. Chem., 2004, 9, 1804–1807 CrossRef.
- A. Dutta, J. Mondal, A. K. Patra and A. Bhaumik, Chem.–Eur. J., 2012, 18, 13372–13378 CrossRef CAS PubMed.
- S. Mathur, M. Veith, H. Shen, S. Hüfner and M. H. Jilavi, Chem. Mater., 2002, 14, 568–582 CrossRef CAS.
- T. Kimura, Chem. Mater., 2005, 17, 337–344 CrossRef CAS.
- T. Kimura, Chem. Mater., 2005, 17, 5521–5528 CrossRef CAS.
- T. Y. Ma, X. Z. Lin, X. J. Zhang and Z. Y. Yuan, Nanoscale, 2011, 3, 1690–1696 RSC.
- T. Y. Ma and Z. Y. Yuan, Dalton Trans., 2010, 39, 9570–9578 RSC.
- S. Dai, M. C. Burleigh, Y. Shin, C. C. Morrow, C. E. Barnes and Z. Xue, Angew. Chem., Int. Ed., 1999, 38, 1235–1239 CrossRef CAS.
- K. Z. Hossain and L. Mercier, Adv. Mater., 2002, 14, 1053–1056 CrossRef CAS.
- J. Huang, M. Ye, Y. Qu, L. Chu, R. Chen, Q. He and D. Xu, J. Colloid Interface Sci., 2012, 385, 137–146 CrossRef CAS PubMed.
- M. M. Natile and A. Glisenti, Chem. Mater., 2002, 14, 3090–3099 CrossRef CAS.
- M. M. Natile and A. Glisenti, Chem. Mater., 2005, 17, 3403–3414 CrossRef CAS.
- J. Y. Luo, M. Meng, X. Li, X. G. Li, Y. Q. Zha, T. D. Hu, Y. N. Xie and J. Zhang, J. Catal., 2008, 254, 310–324 CrossRef CAS PubMed.
- H. Liang, H. Sun, A. Patel, P. Shukla, Z. H. Zhu and S. Wang, Appl. Catal., B, 2014, 127, 330–335 CrossRef PubMed.
- Y. Hardjonoa, H. Q. Sun, H. Y. Tian, C. E. Buckley and S. B. Wang, Chem. Eng. J., 2011, 174, 376–382 CrossRef PubMed.
- P. Shukla, H. Q. Sun, S. B. Wang, H. M. Ang and M. O. Tade, Sep. Purif. Technol., 2011, 77, 230–236 CrossRef CAS PubMed.
- P. Shukla, S. Wang, K. Singh, H. M. Ang and M. O. Tadé, Appl. Catal., B, 2010, 99, 163–169 CrossRef CAS PubMed.
- E. Saputraa, S. Muhammada, H. Sun, H. M. Ang, M. O. Tadé and S. Wang, Catal. Today, 2012, 190, 68–72 CrossRef PubMed.
- P. Shukla, H. Sun, S. Wang, H. M. Ang and M. O. Tadé, Catal. Today, 2011, 175, 380–385 CrossRef CAS PubMed.
- S. Muhammad, E. Saputra, H. Sun, J. C. Izidoro, D. A. Fungaro, H. M. Ang, M. O. Tadé and S. Wang, RSC Adv., 2012, 2, 5645–5650 RSC.
- Y. Wang, H. Sun, H. M. Ang, M. O. Tadé and S. Wang, Chem. Eng. J., 2014, 245, 1–9 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra15032e |
| This journal is © The Royal Society of Chemistry 2015 |