DOI:
10.1039/C4RA05689B
(Paper)
RSC Adv., 2014,
4, 38111-38118
Enhanced photovoltaic performance of silver@titania plasmonic photoanode in dye-sensitized solar cells†
Received
13th June 2014
, Accepted 12th August 2014
First published on 13th August 2014
Abstract
In the present investigation, silver@titania (Ag@TiO2) plasmonic nanocomposite materials with different Ag content were prepared using a simple one-step chemical reduction method and used as a photoanode in high-performance dye-sensitized solar cells. Transmission electron microscopic images revealed the uniform distribution of ultra-small Ag nanoparticles with a particle size range of 2–4 nm on the TiO2 surface. The incorporation of Ag on the TiO2 surface significantly influenced the optical properties in the region of 400–500 nm because of the surface plasmon resonance effect. The dye-sensitized solar cells (DSSCs) assembled with the Ag@TiO2-modified photoanode demonstrated an enhanced solar-to-electrical energy conversion efficiency (4.86%) compared to that of bare TiO2 (2.57%), due to the plasmonic effect of Ag. In addition, the Ag nanoparticles acted as an electron sink, which retarded the charge recombination. The influence of the Ag content on the overall efficiency was also investigated, and the optimum Ag content with TiO2 was found to be 2.5 wt%. The enhanced solar energy conversion efficiency of the Ag@TiO2 nanocomposite makes it a promising alternative to conventional photoanode-based DSSCs.
1. Introduction
Renewable energy sources are the most important approaches for and signify an important method for gaining independence from fossil fuels. Utilizing solar energy is certainly one of the most viable ways to solve the world's energy crisis. Dye-sensitized solar cells (DSSCs) have emerged as promising candidates for harnessing solar power because of their low cost, flexibility, ease of production, relatively high energy conversion efficiency, and low toxicity to the environment.1 Since first being introduced by Gratzel and co-workers in 1991, many strategies have been employed to achieve high-performance DSSCs, including novel counter electrodes, electrolytes, dyes, and semiconductor photoanode materials. Among these, the photoanode plays a crucial role in determining the cell performance. So far, titanium dioxide (TiO2)-based material is one of the most promising materials for a DSSC due to its low cost, abundance, nontoxicity, safety, large surface area for maximum dye uptake and matched energy and band structure.2,3 However, the major drawback associated with the use of TiO2 is its random electron transport, which will cause the electron–hole recombination process and hence affect the overall performance.4,5
There is an active search to overcome the deficiency of TiO2-based DSSCs such as through surface modification with metal nanoparticles, doping of metals and non-metals, semiconductor coupling, and hybridizing with a carbon materials.6–9 In the present decade, surface of TiO2 has been modified with noble metal nanoparticles such as silver (Ag), with the aim of improving the efficiency of a DSSC. The Ag nanoparticles play dual roles in the DSSC performance, including the enhancement of the absorption coefficient of the dye and optical absorption due to surface plasmonic resonance.10–12 Moreover, they act as an electron sink for photoinduced charge carriers, improve the interfacial charge transfer process, and minimize the charge recombination, thereby enhancing the electron transfer process in a DSSC.13–15 Hence, the performance of a DSSC with Ag@TiO2 plasmonic nanocomposite material-modified photoanodes has been actively investigated.13–15
To make use of economically viable Ag to boost the DSSC performance, it is essential to control both the size and distribution of the nanoparticles on the TiO2 surface. Two method are commonly employed for Ag–TiO2 nanocomposite preparation: (i) a two-step method involving the chemical and physical adsorption of preformed Ag nanoparticles on the TiO2 surface16–18 and (ii) the photoreduction of Ag on the TiO2 surface.19,20 However, these synthetic methods are ineffective because of the aggregation of Ag nanoparticles in the first method and the difficulty controlling the size of the Ag nanoparticles in the later one.
In the present study, we successfully developed a facile synthesis method to prepare uniformly distributed Ag nanoparticles deposited on TiO2 using a simple one-step chemical reduction method without adding any stabilizer or surfactant for DSSC application. The as-prepared Ag@TiO2 plasmonic nanocomposites were characterized using various suitable analytical techniques and used as photoanodes in the DSSCs. The Ag@TiO2 plasmonic nanocomposite-modified photoanode showed an enhanced solar energy conversion efficiency compared to that of a bare TiO2-based DSSC. The effect of the Ag content on the DSSC performance was also investigated. There are many positive aspects of the Ag-modified TiO2, including the synergetic interaction of the Ag nanoparticles on the TiO2 surface, surface plasmon resonance effect, reduction of the band gap, and enhancement of the charge transfer process. These multifunctional properties of the prepared Ag@TiO2 plasmonic nanocomposite will lead to superior performance in a DSSC.
2. Experimental methods
2.1. Materials and characterization techniques
Titanium dioxide (P25) was purchased from Acros Organics. Silver nitrate (AgNO3) and sodium borohydride (NaBH4) were purchased from Merck. Indium tin oxide (ITO) conducting glass slides (7 Ω sq−1) were purchased from Xin Yan Technology Limited, China. The N719 (Ruthenizer 535-bisTBA) dye and Iodolyte Z-100 were received from Solaronix. The crystalline phase of the samples was studied via X-ray diffraction (XRD; D5000, Siemens), using copper Kα radiation (λ = 1.5418 Å). The morphologies of the prepared samples were examined using a Hitachi-SU 8000 field emission scanning electron microscope and JEOL JEM-2100 F high-resolution transmission electron microscope. The absorption spectra were assessed using a Thermo Scientific Evolution 300 UV-vis absorption spectrophotometer. Photoluminescence and Raman spectra were collected using a Renishaw inVia 2000 system with an argon ion laser emitting at 325 and 532 nm, respectively. X-ray photoelectron spectroscopy (XPS) measurements were performed using synchrotron radiation from beamline no. 3.2 at the Synchrotron Light Research Institute, Thailand.
2.2. Synthesis of Ag@TiO2 nanocomposite materials
The Ag@TiO2 nanocomposite materials were prepared using a simple one-step chemical reduction method. Briefly, 500 mg of TiO2 were added to aqueous solutions that contained different amounts of AgNO3 (1, 2.5, 5, 10, and 20 wt%). Each mixture was vigorously stirred for 30 min at room temperature. The reduction of Ag+ was carried out by the drop-wise addition of NaBH4 until the color changed to greenish yellow. The appearance of this greenish yellow color indicated the formation of the Ag@TiO2 nanocomposite, and the solution was continually stirred for another 30 min. The Ag@TiO2 nanocomposite was collected and washed with distilled water and ethanol several times by centrifugation. Finally, the product was dried in an oven at 60 °C and stored under a dark condition.
2.3. Fabrication of Ag@TiO2-modified photoanode
Ag@TiO2 modified photoanodes were fabricated using the following procedure. Initially, 300 mg of the Ag@TiO2 nanocomposite was mixed in an ethanolic solution and stirred for 30 min. A 0.1 M quantity of TTIP was slowly introduced into the above reaction mixture and stirred until a homogenous solution was obtained. Finally, the Ag@TiO2 nanocomposites were coated on a conducting side of the ITO using the doctor-blade technique with the aid of scotch-3M tape and the thickness of the film was ∼12 μm. In order to obtain a stable photoanode, the film was dried at room temperature, sintered at 150 °C for 30 min in a muffle furnace, and then allowed to cool naturally to room temperature.
2.4. Fabrication of DSSCs and evaluation of their performances
The prepared Ag@TiO2 plasmonic nanocomposite photoanodes were immersed in a ethanolic solution of 0.3 mM N719 (Ruthenizer 535-bisTBA) dye for 24 h at room temperature under a dark condition. The dye-adsorbed plasmonic photoanode was withdrawn from the solution and immediately but gently cleaned with ethanol. A platinum-sputtered ITO was placed on a dye-absorbed photoanode, and they were clamped firmly together. A redox electrolyte (Iodolyte Z-100, Solaronix) solution was introduced into the cell assembly by capillary action. An active area of 0.5 cm2 was used to measure the cell performance. A 150 W Xenon arc lamp (Newport, Model 69907) containing a simulated AM 1.5G filter with a manual shutter was used as a light source throughout the experiments. Prior to testing the photovoltaic parameter, an Avaspec-2048 fiber optic spectrophotometer was used to measure the light illumination intensity. The photocurrent signal measurements (J–V and J–T curves) and electrochemical properties of the fabricated DSSCs were studied by using a computer-controlled VersaSTAT 3 Electrochemical Workstation (Princeton Applied Research, USA).
3. Results and discussion
3.1. Optical properties of Ag@TiO2 nanocomposite materials
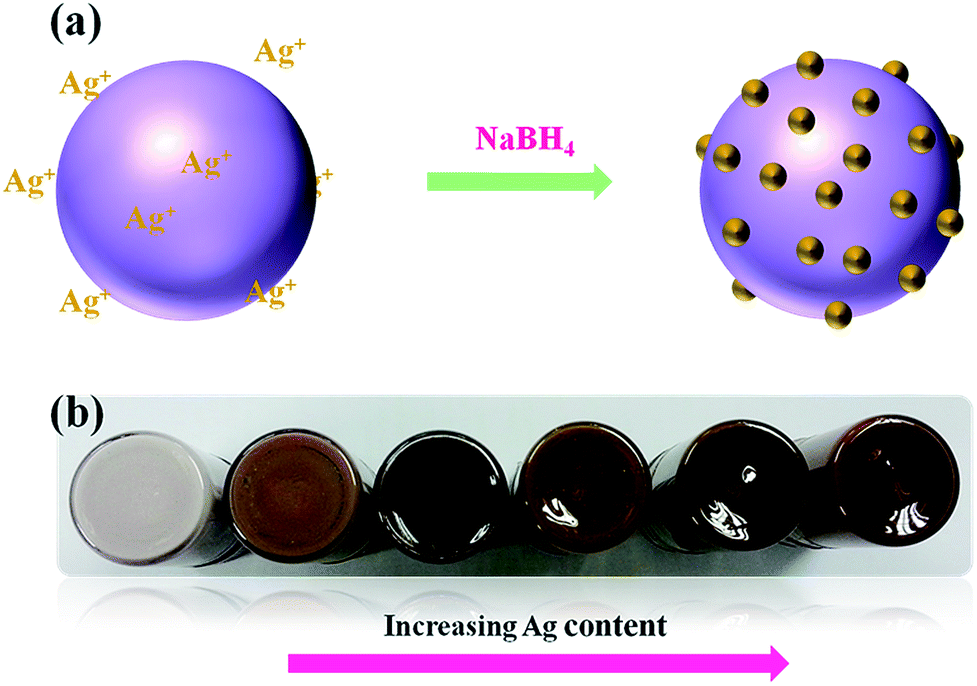
In the present synthetic method, the formation of the Ag@TiO2 nanocomposite takes place through the adsorption of Ag+ ions on the TiO2 surface, followed by the chemical reduction of Ag+ by NaBH4 at room temperature in the absence of any stabilizer and surfactant (Fig. 1a). The physical appearances of the Ag@TiO2 with different quantities of Ag (wt%) are shown in Fig. 1b, shows that the nanocomposite becomes darker in color with increasing Ag content.
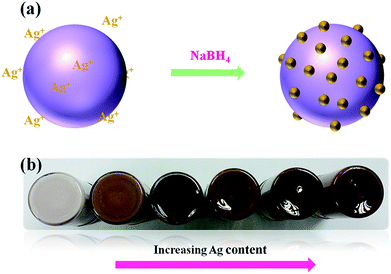 |
| | Fig. 1 (a) Schematic representation of formation of Ag on TiO2 surface and (b) physical appearance of as-prepared Ag@TiO2 nanocomposites with different Ag content. | |
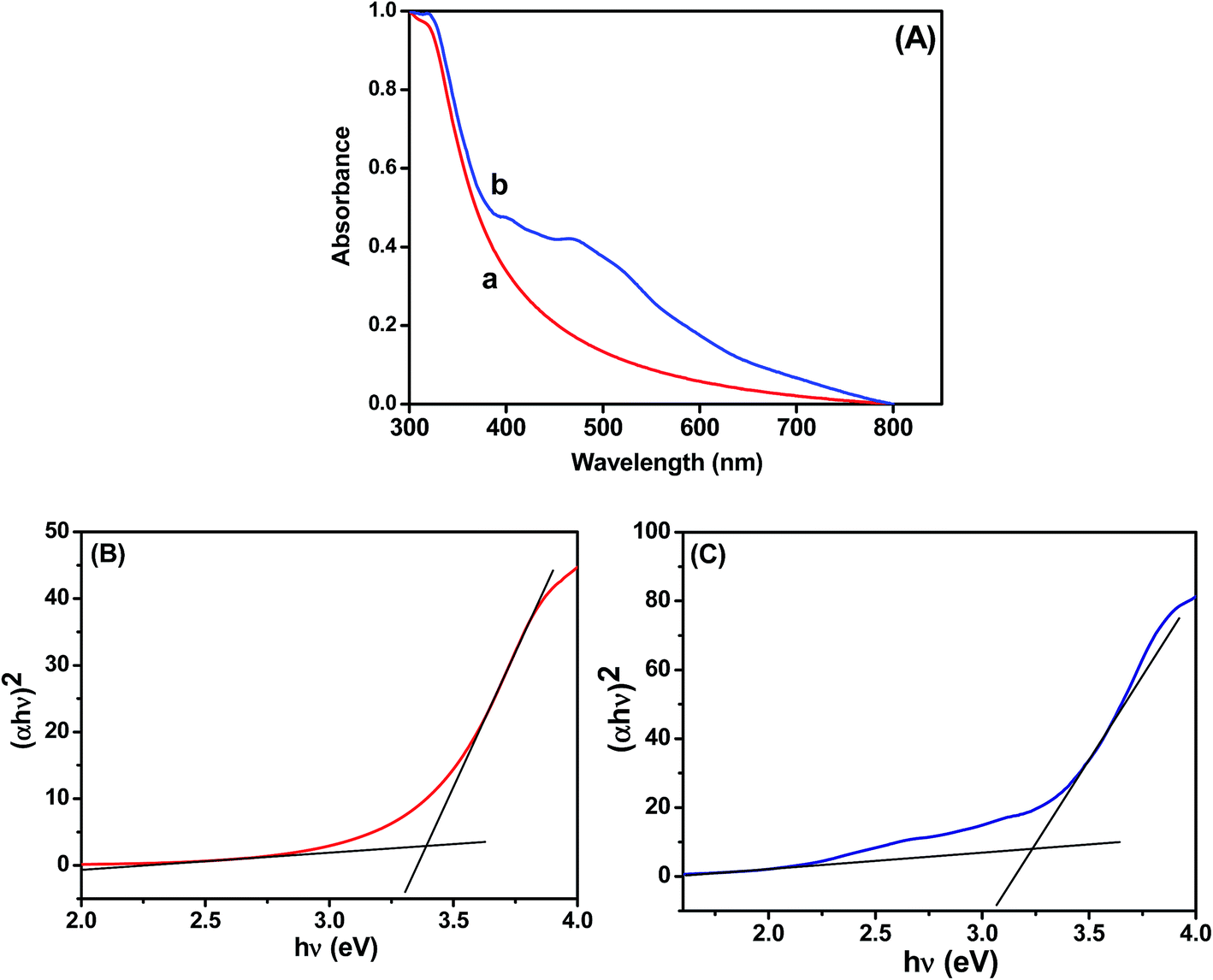
The UV-vis absorption spectra of the TiO2 and Ag@TiO2 were recorded and are shown in Fig. 2. The TiO2 did not show any absorbance in the visible region (Fig. 2A) because of the wide band gap (∼3.2 eV). The deposition of Ag on the TiO2 surface significantly influenced the absorption in the visible regions of 450 and 500 nm, which was due to the surface plasmon resonance (SPR) band of Ag nanoparticles.19,21 A considerable shift in the adsorption edge toward the visible region was also observed for the Ag@TiO2 sample. The presence of Ag nanoparticles significantly influenced the visible light absorption properties of TiO2. The band-gap energy (Ebg) of the prepared TiO2 and Ag@TiO2 were calculated using a well-known Tauc's plot method.22,23 The relations of (αhν)2versus hν for the TiO2 and Ag@TiO2 are shown in Fig. 2B and C. It can be observed that the band-gap energy values of TiO2 decreased from 3.36 eV to 3.22 eV with the addition of Ag nanoparticles. This may have been due to the presence of Ag, which decreased the absorbance band edge of TiO2 close to the visible region.
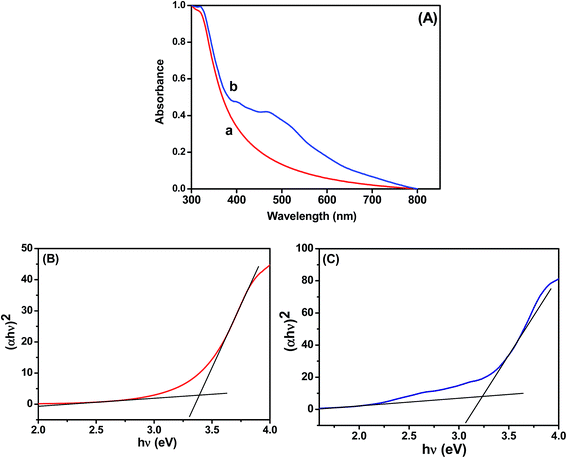 |
| | Fig. 2 (A) Absorption spectra of (a) TiO2 and (b) Ag@TiO2. Plots of (αhν)2versus hν obtained for (B) TiO2 and (C) Ag@TiO2. | |
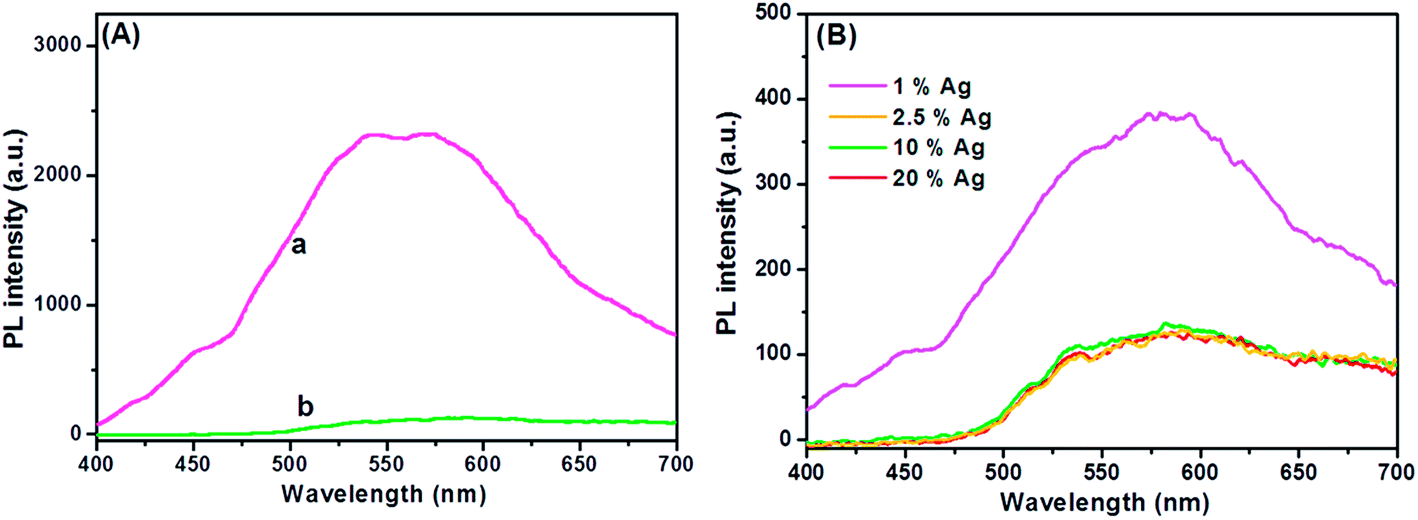
Understanding the charge recombination process of a photoanode material is crucial because it can significantly influence the photovoltaic performance of a DSSC. The TiO2 will absorb incident photons with sufficient energy equal to or higher than the band-gap energy. This will produce photoinduced charge carriers (h+⋯e−), and the recombination of photoinduced electrons and holes will release energy in the form of photoluminescence. Hence, a lower PL intensity indicates less charge recombination. The TiO2 showed a broad and high PL intensity at around 580 nm due to the high photoinduced charge carrier recombination, whereas the PL intensity was minimized upon the addition of Ag on the TiO2 surface (Fig. 3A). This was mainly attributed to the formation of the Schottky barrier at the Ag–TiO2 interface, which could act as an electron sink to efficiently prevent the electron–hole recombination process.24 The Ag@TiO2 with 2.5 wt% Ag showed the lowest PL emission intensity, which indicated the least electron–hole recombination compared to Ag contents of 1, 10, and 20 wt% on the TiO2 (Fig. 3B).
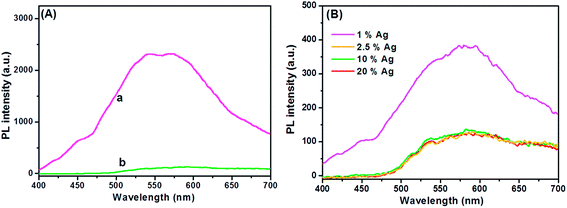 |
| | Fig. 3 (A) Photoluminescence spectra of (a) TiO2 and (b) Ag@TiO2 nanocomposites under 325 nm laser excitation. (B) Photoluminescence spectra of Ag@TiO2 nanocomposites with different Ag contents under 325 nm laser excitation. | |
3.2. Crystalline properties of Ag@TiO2 nanocomposite materials
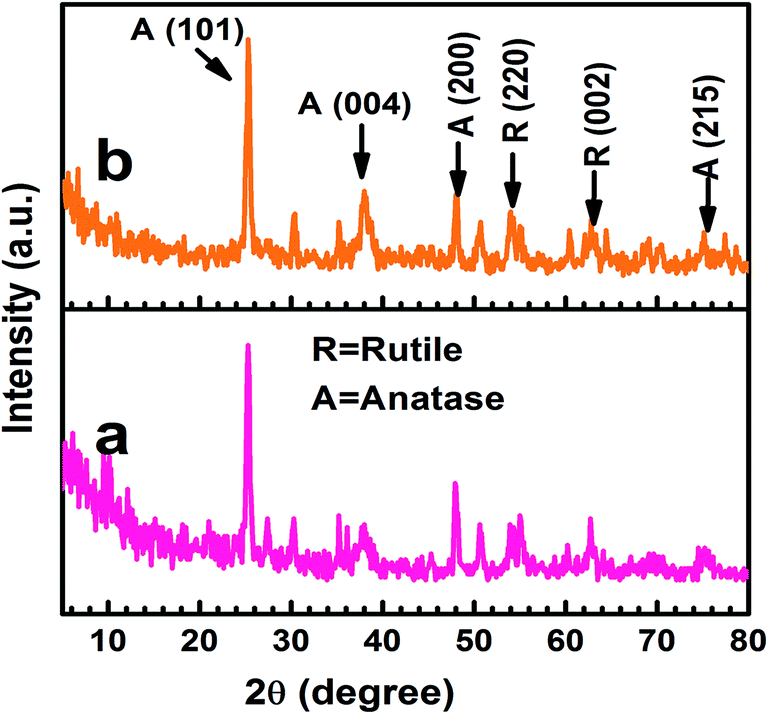
The X-ray diffraction patterns indicated that the TiO2 and Ag@TiO2 were composed of mixed anatase and rutile phases (Fig. 4), which was in good agreement with the reference patterns of JCPDS card no. 83-2243 and 21-1276, respectively. The diffraction peaks observed at the 2θ values of 25.42°, 38.68°, 48.15°, and 74.4° corresponded to the anatase phase of TiO2 and were assigned to the (101), (004), (200), and (215) crystallographic planes, respectively. In contrast, the peaks at the 2θ values of 54.08° and 63.81° agreed well with the rutile phase of TiO2 and were are assigned to the (220) and (002) crystallographic planes, respectively. The crystallographic peaks due to the Ag overlapped with those for the rutile phase of the TiO2. Hence, the peaks were indistinguishable in Ag@TiO2.
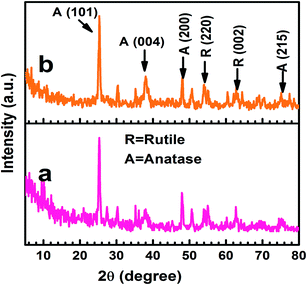 |
| | Fig. 4 X-ray diffraction patterns of (a) TiO2 and (b) Ag@TiO2 nanocomposites. | |
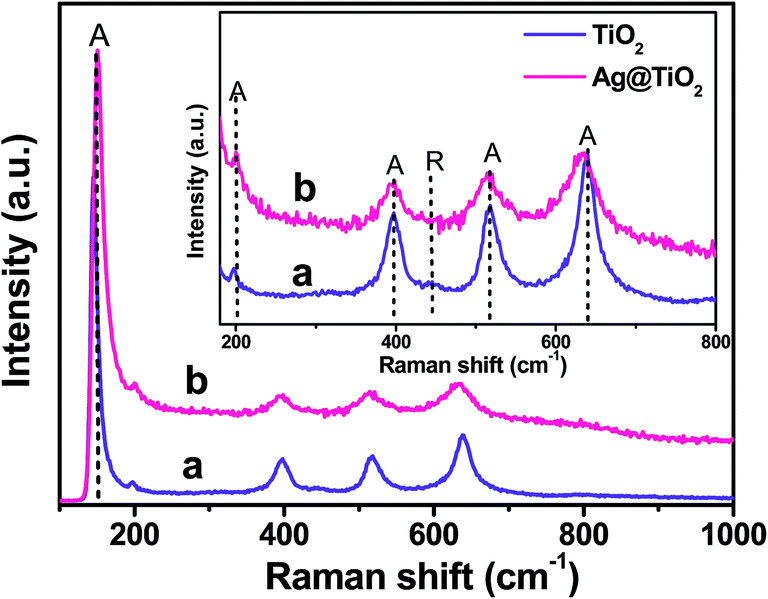
To further evaluate the phase identification of the TiO2 and Ag@TiO2, Raman spectroscopy was performed in the range of 100–1000 cm−1, and the results are shown in Fig. 5. The anatase TiO2 phase was observed at 153, 198, 396, 516, and 637 cm−1, whereas the rutile TiO2 phase was detected at 443 cm−1.25–27 This clearly indicated that the TiO2 and Ag@TiO2 nanoparticles contained a mixture of anatase and rutile phases. No signals related to Ag particles were identified for the samples because of the relatively low concentration of Ag loaded onto the TiO2 and its weak Raman scattering. An interesting observation is that the peak intensities were reduced by the presence of Ag, but the position of the Raman signal remained the same and was broadened. This indicated that the interaction between the Ag and TiO2 affected the Raman resonance of the TiO2.28 This observation showed the successful deposition of Ag on the TiO2 surface without any phase transition.22,27
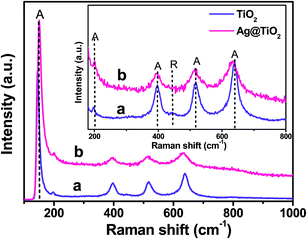 |
| | Fig. 5 Raman spectra of (a) TiO2 and (b) Ag@TiO2 nanocomposites with inset showing the anatase and rutile features of TiO2. | |
3.3. XPS analysis of Ag@TiO2 nanocomposite materials
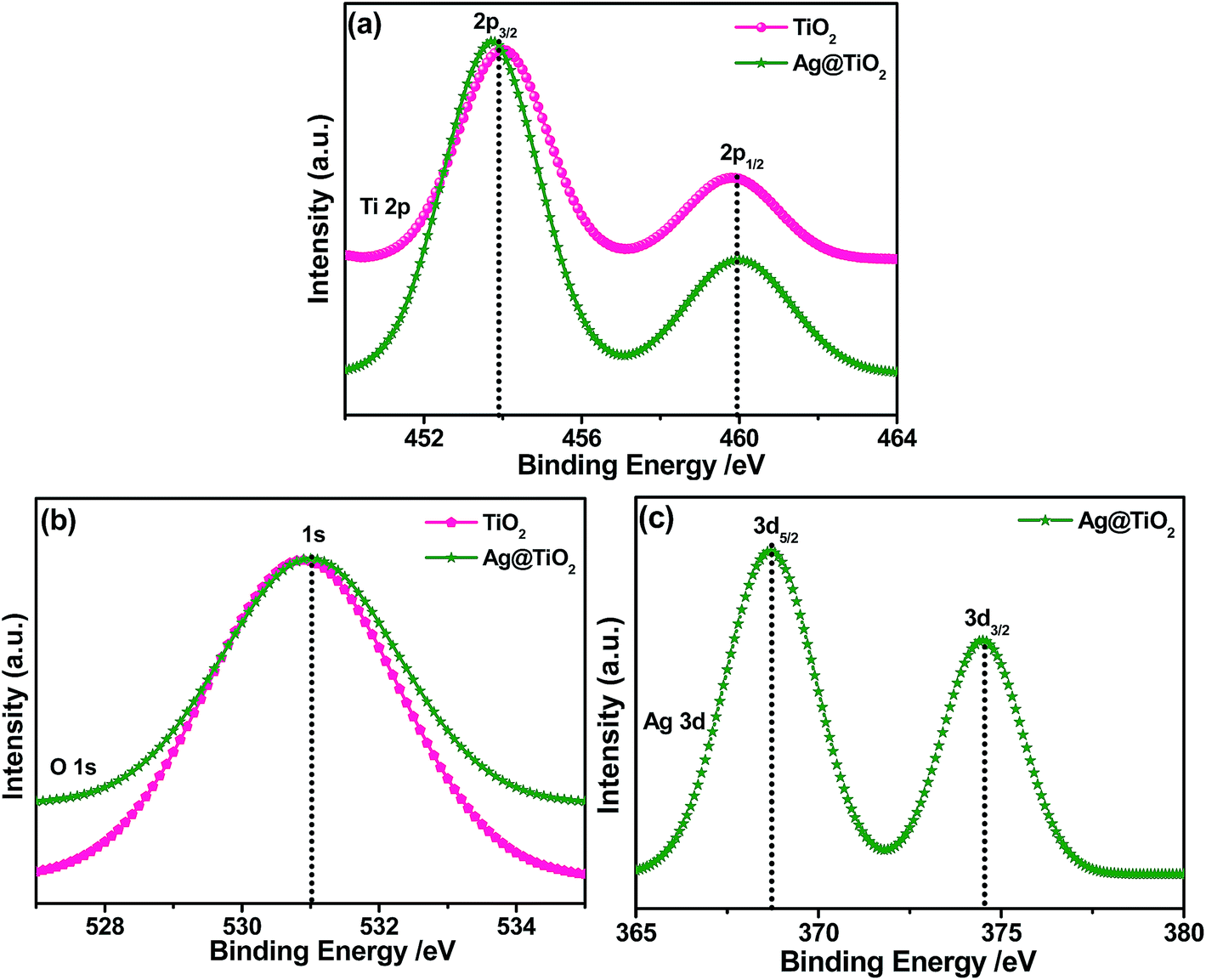
The XPS spectra of the TiO2 and Ag@TiO2 were recorded to understand their chemical nature and are shown in Fig. 6. Fig. 6A shows the Ti 2p core level spectra for both samples, in which two peaks are observed at 454.1 and 459.9 eV corresponding to the binding energies of the Ti 2p3/2 and Ti 2p1/2 core levels due to the presence of the Ti(IV) state.29 After the deposition of the Ag nanoparticles, it is obviously observed that the Ti 2p peak was shifted to lower binding energies due to its surrounding chemical environment.30Fig. 6B shows the O 1s spectra of the TiO2 and Ag@TiO2, and the binding energy of the O 1s state of the samples is located at 530.9 eV, which is assigned to the bulk oxides (O2−) in the P25 lattice. The O 1s is slightly shifted to higher binding energies which indicate the increase in electron density around O atoms due to the interaction of residue precursor with TiO2.21 The binding energy found for the Ag 3d5/2 and Ag 3d3/2 levels are 367.5 and 373.5 eV, respectively (Fig. 6C), with a peak separation of 6 eV due to the metallic silver.21 The XPS analysis provided support for the existence of elements such as Ti, O, and Ag in the nanocomposite materials.
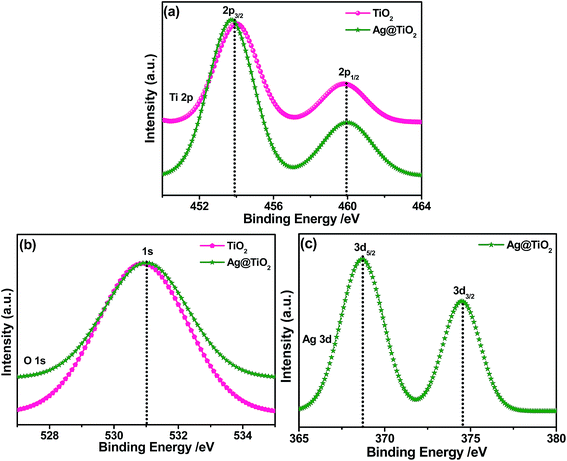 |
| | Fig. 6 XPS spectra of TiO2 and Ag@TiO2 and their corresponding (a) Ti 2p (b) O 1s, and (c) Ag 3d core-level spectra. | |
3.4. Morphological studies of Ag@TiO2 nanocomposite materials
The microscopic morphologies of the as-prepared samples were studied using FESEM, TEM, and HRTEM. Fig. S1a† shows the FESEM results for TiO2, which appear to be spherical with a uniform size. Upon the addition of Ag, no significant change in morphology was observed for the film (Fig. S1b†).
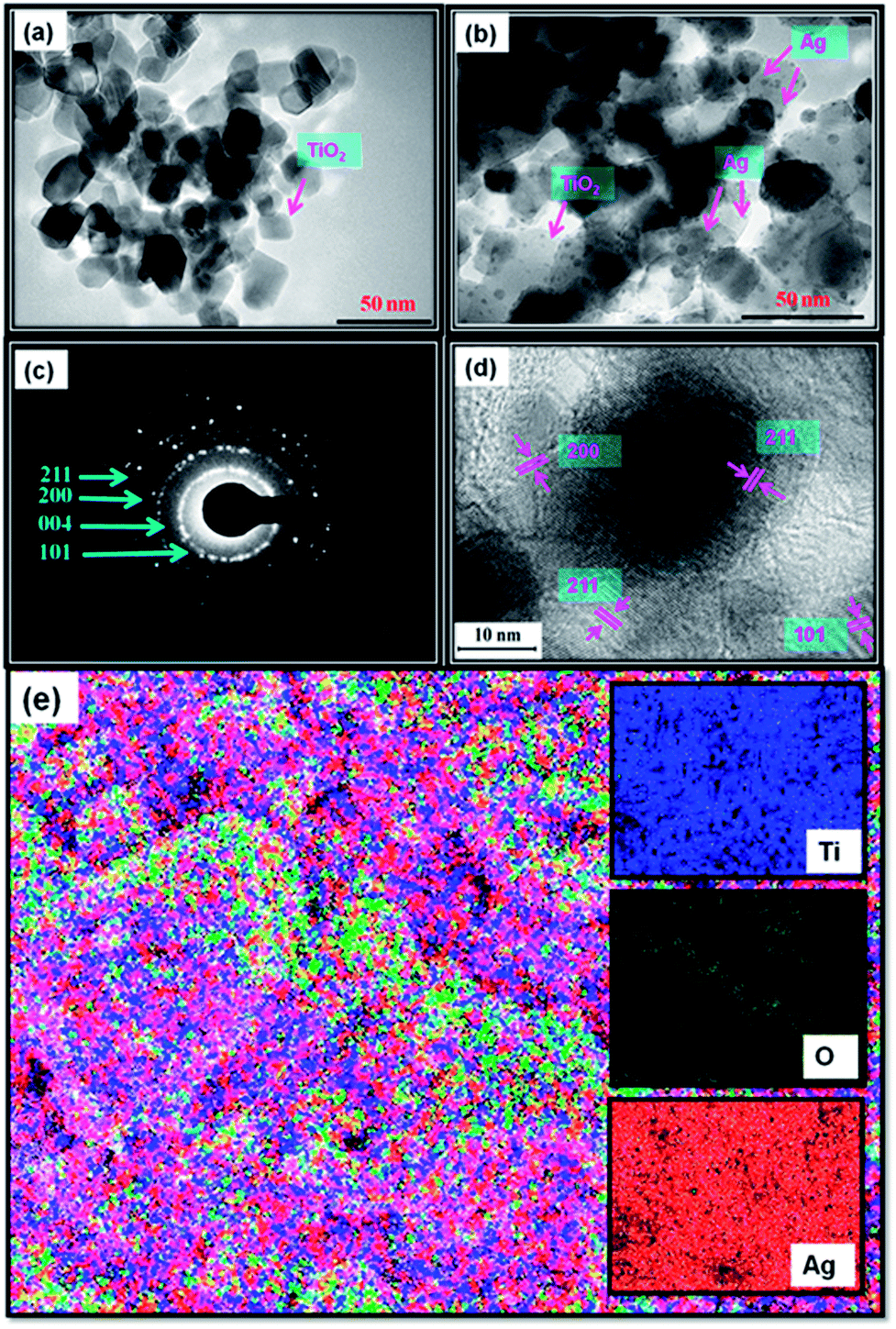
The EDX analysis results are shown in Fig. S1c,† which shows Ti, O, C, and Ag. The C peak found in the EDAX spectrum is a result of carbon tape. Further, TEM images of TiO2 and Ag@TiO2 (2.5 wt% Ag) were also recorded and are shown in Fig. 7a and b, respectively. The TEM image shows that the Ag@TiO2 nanoparticles are spherical in shape, with the TiO2 particles having a size range of 20–25 nm. Fig. 7b clearly shows the deposition of distributed spherical and smaller Ag nanoparticles (2–4 nm) on the surface of TiO2. Fig. 7c depicts the selected area electron diffraction (SAED) pattern of the nanocrytalline TiO2 particles. This pattern clearly reveals bright concentric rings, which are due to the diffraction from the (211), (200), (004), and (101) planes of anatase TiO2. The lattice resolved HRTEM image of the Ag@TiO2 (Fig. 7d) shows d-spacing values for the lattice fringes of 2.28 Å and 2.50 Å, which correspond to the (200) and (101) rutile planes of TiO2, respectively; whereas the interplanar spacing of 1.89 Å was assigned to the (200) plane of anatase TiO2. In Fig. 7e, the areas of bright contrast on the element maps correlate with the Ti, O, and Ag signal maps.
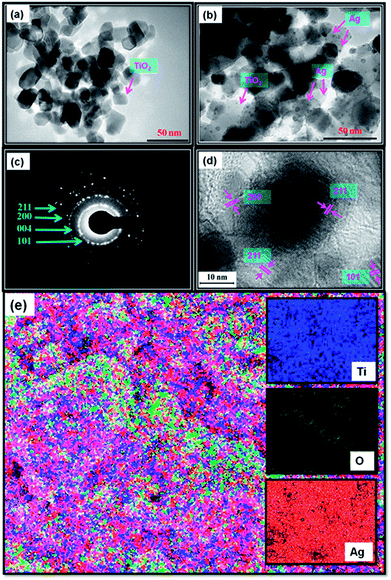 |
| | Fig. 7 TEM images of (a) TiO2 and (b) Ag@TiO2 nanocomposites, (c) SAED pattern, (d) lattice-resolved TEM image, and (e) elemental mapping of Ag@TiO2 nanocomposite. | |
3.5. Photovoltaic performances of Ag@TiO2 plasmonic nanocomposite-modified photoanode-based DSSCs
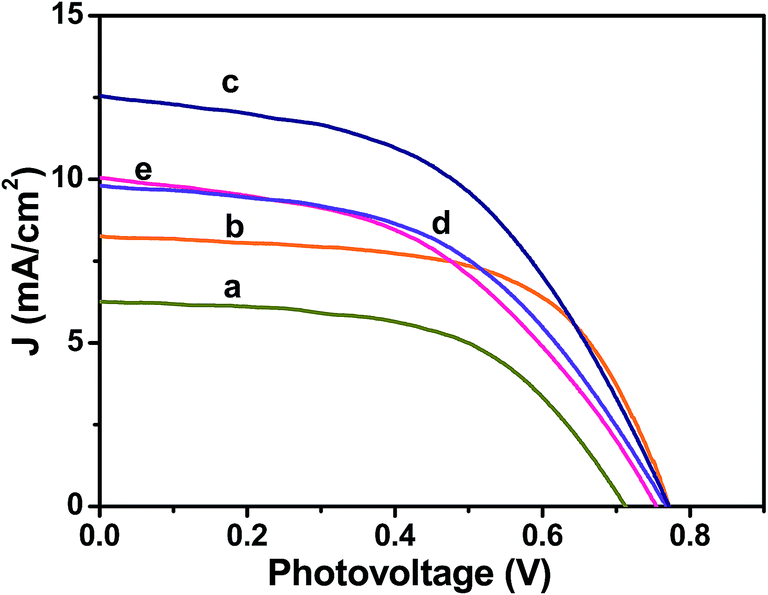
The photovoltaic performances of the Ag@TiO2 plasmonic nanocomposite-modified photoanode-based DSSCs with different Ag contents were evaluated under simulated solar AM 1.5G irradiation. Their obtained photocurrent density and photovoltage (J–V) curves are shown in Fig. 8, and their evaluated photovoltaic parameters are listed in Table 1. The Ag@TiO2 plasmonic photoanode (2.5 wt% of Ag) showed a higher efficiency (4.86%) than the unmodified TiO2 (2.57%). The enhanced photovoltaic performance may have been due to the plasmonic effect and rapid interfacial charge transfer that arose from the Ag nanoparticles on the TiO2. The optimization of Ag on TiO2 is essential from the economic and high-performance perspectives for a DSSC. Although increasing the Ag content on the TiO2 surface beyond 2.5 wt% showed a decrease in the conversion efficiency to 3.59%, the observed results clearly revealed that the conversion efficiency of a DSSC was increased with an increase in the Ag content of the photoanode until it reached a maximum of 2.5%. Then, a further increase in the Ag content eventually led to the decrease in conversion efficiency (Fig. 8a and Table 1). The decrease in the efficiency at high Ag loading is due to the free standing/excess Ag in the composite may oxidized to Ag(I)13,31 and eroded by the electrolyte.13 The oxidation of the Ag will act as new recombination centre, thus reducing the number of the charge carrier led to decrease in the Jsc and Voc. Consequently, the overall conversion efficiency of the DSSC would have deteriorated. Moreover, the addition of Ag more than 2.5 wt% might result in decrease of the active surface area of TiO2 interacted with the dye molecules. Hence, the recombination between the electrons and holes will increase leads to decrease of Jsc value. Consequently, the overall conversion efficiency of the DSSC would have deteriorated. The relationships between the photovoltaic parameters and Ag content on the TiO2 surface are represented in Fig. S2(a–c)† for better understanding.
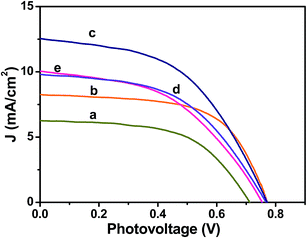 |
| | Fig. 8 Photocurrent density–photovoltage (J–V) curves obtained for Ag@TiO2 nanocomposite thin films with (a) 0, (b) 1, (c) 2.5, (d) 10, and (e) 20 wt% of Ag content under 100 mW cm−2 simulated AM 1.5G solar light irradiation. | |
Table 1 Photovoltaic parameters of Ag@TiO2 nanocomposite-modified photoanode-based DSSCs with different Ag contentsa
| Ag (wt%) |
J
sc (mA cm−2) |
V
oc (V) |
J
max (mA cm−2) |
V
max (V) |
FF |
η (%) |
|
The DSSC performance was evaluated under 100 mW cm−2 simulated AM 1.5G solar light irradiation. Jsc: short-circuit current density; Voc: open-circuit voltage; Jmax: maximum photocurrent density; Vmax: maximum photovoltage; FF: fill factor; η: power conversion efficiency. Area of the cell electrode was 0.5 cm2.
|
| 0 |
6.71 |
0.71 |
5.24 |
0.49 |
0.54 |
2.57 |
| 1 |
8.17 |
0.77 |
6.63 |
0.60 |
0.63 |
3.98 |
| 2.5 |
12.19 |
0.77 |
9.34 |
0.52 |
0.52 |
4.86 |
| 10 |
9.93 |
0.77 |
7.65 |
0.49 |
0.49 |
3.75 |
| 20 |
9.96 |
0.75 |
7.68 |
0.45 |
0.48 |
3.59 |
3.6. Electrochemical behaviours of Ag@TiO2 plasmonic photoanode-based DSSCs
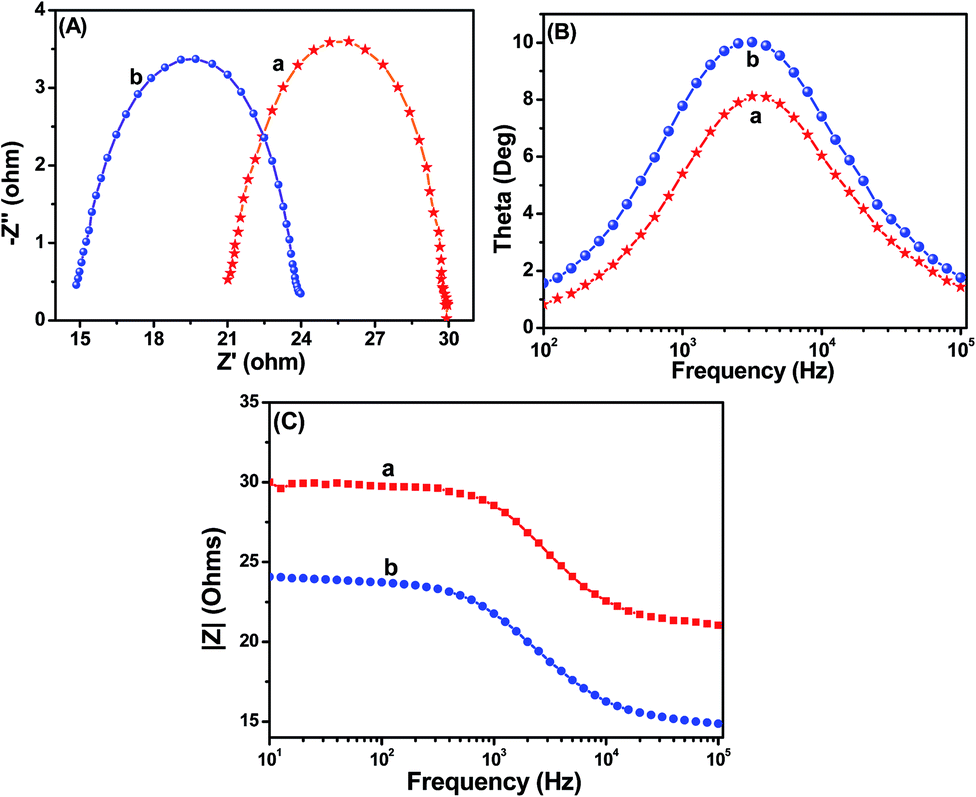
In order to gain deeper insight into the interfacial charge transfer process within the fabricated DSSC, the electrochemical impedance spectra (EIS) were recorded in a frequency range between 0.01 Hz and 100 kHz, and are shown in Fig. 9. A well-defined semicircle in the middle-frequency region can be observed for the TiO2 and Ag@TiO2 based DSSCs. The intersection of a high-frequency semicircle at the real axis represents the equivalent series resistance of the device (Rs); the arc in the middle-frequency range between 1 and 1000 Hz represents the charge transfer resistance (Rct) between the dye-adsorbed photoanode and the electrolyte interface.16,18 From the Nyquist plot (Fig. 9A), the Rs values for the TiO2 and Ag@TiO2 nanocomposite-based DSSCs are 21.03 Ω and 23.96 Ω, respectively. The Rct value increased from 8.77 Ω to 9.01 Ω after the addition of the Ag. This increase in the Rct value could affect the open-circuit voltage (Voc) and fill factor (FF) of the DSSC device. Therefore, the origin of the higher Jsc in the Ag@TiO2 is expected to arise from the device resistance (Rs) and charge transport dynamics determined by the electron lifetime (τn) and Rct. Based on the Bode phase plots in Fig. 9B, the frequency was apparently shifted to a lower frequency region with the addition of Ag. The maximum frequencies (ωmax) in the middle-frequency region of the Bode plots for TiO2 and Ag@TiO2 were 2511.89 Hz and 1995.26 Hz, respectively. Because ωmax is inversely associated with the electron lifetime τn = 1/(2πf),32,33 a decrease in ωmax indicates a reduced rate for the charge-recombination process in the DSSC. Electrons with longer τn values will survive from the recombination. Therefore, it will be characterized by a larger Rct.
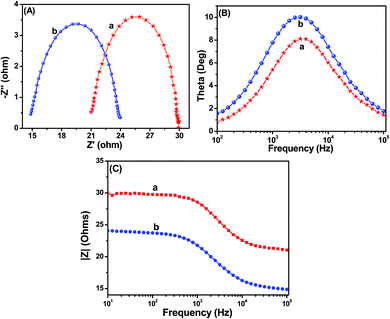 |
| | Fig. 9 (A) Nyquist plots, (B) Bode angle phase plots, and (C) Bode amplitude plots obtained for (a) TiO2 and (b) Ag@TiO2 nanocomposite (2.5 wt% of Ag content)-based DSSCs. | |
Furthermore, Table 2 and Fig. S3† summarize the results of the Nyquist plot. The Ag@TiO2 exhibited a faster electron transport time (τs = RsCμ)32,34,35 than the TiO2. Hence its electron lifetime (τn = RctCμ)32,34,35 was significantly increased and survived from the recombination. The photovoltaic performance of the DSSC is clearly reflected by the charge collection efficiency (ηc)32,34,35 derived from ηc = (1 + Rs/Rct)−1. Eventually, the charge collection efficiency was significantly increased with the addition of Ag. We can conclude that because of the longer τn and larger Rct, the devices fabricated using Ag@TiO2 showed improved Jsc values compared to TiO2.
Table 2 Electrochemical parameters of TiO2 and Ag@TiO2 nanocomposite-based DSSCsa
| Photoanode |
R
s (Ω) |
R
ct (Ω) |
C
μ (μF) |
τ
s (ms) |
τ
n (ms) |
η
c (%) |
|
The electrochemical impedance spectra (EIS) were recorded at an applied bias of −0.7 V in the frequency range of 0.01 Hz to 100 kHz. Rs: device resistance; Rct: charge transfer resistance; Cμ: chemical capacitance; τs: electron transport time; τn: electron lifetime; ηc: charge collection efficiency.
|
| TiO2 |
21.13 |
8.77 |
7.22 |
0.15 |
0.06 |
29 |
| Ag@TiO2 |
23.93 |
9.01 |
8.85 |
0.13 |
0.08 |
38 |
3.7. Operation principle of Ag@TiO2 plasmonic photoanode-based DSSC
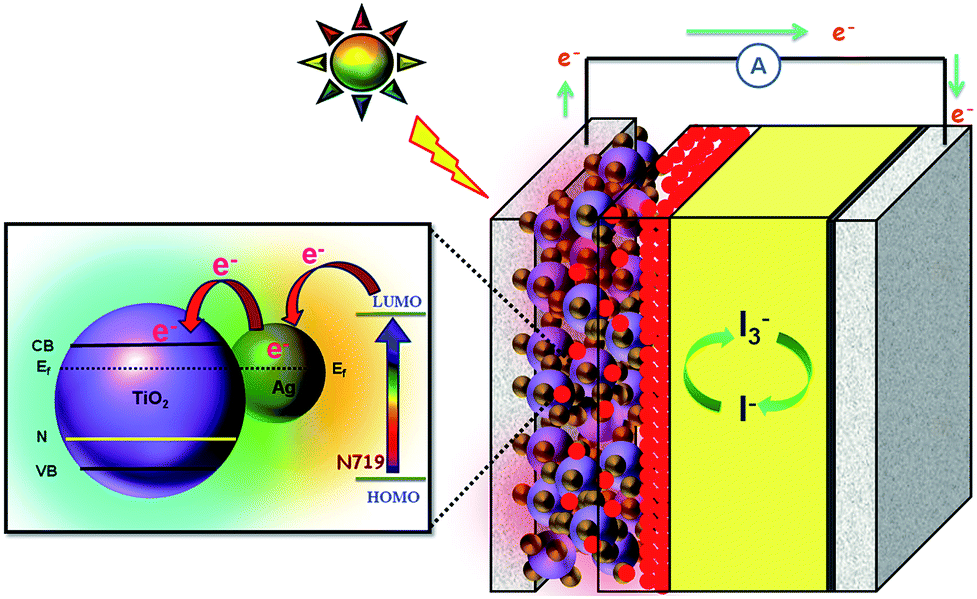
The operation principle of the DSSC based on the Ag@TiO2-modified photoelectrode under illumination is shown in Fig. 10. During light irradiation, the dye absorbs incident light and promotes electrons to the excited state. An excited electron is injected into the conduction band of the TiO2 nanoparticles. The dye is then oxidized by receiving the electron from the electrolyte through the redox system, and is ready to be used again. The electrolyte itself will regenerate via the platinum counter-electrode by an electron passing through the external circuit. In our study, the Ag deposited onto the TiO2 surface not only acted as an electron sink for the photoinduced charge carriers but could also be used as a scattering element for plasmonic scattering to trap the light and near field coupled with the dye molecules.36 This will eventually improve the optical absorption of the dye, resulting in a significant enhancement of the photocurrent (Fig. 10). Whenever the TiO2 comes to contact with Ag, both will undergoes Fermi level equilibration to each other, resulted in the formation of Schottky barrier and thus led to large numbers of electron are accumulated at the surface of the metal (Ag) nanoparticles. This accumulation of electrons on the Ag nanoparticles shifted the position of the Fermi level closer to the conduction band of TiO2.37 The Schottky barrier formed between the Ag and TiO2 helps to flow the electrons from the Ag to TiO2 conduction band via rapid interfacial charge transfer process and were collected by the current collector (ITO), thus improve the photocurrent generation under irradiation.38 Once the electrons being transfer to the conduction band of TiO2, the photo-excited electrons from the dye, N719, will start to accumulate on Ag surface again. Furthermore, the formation of Schottky barrier at the Ag–TiO2 interface, which could act as an electron sink, also will help to prevent the electron–hole recombination process.24 The similar report also available for the metal-semiconductor (Ag–TiO2) photoanode sensitized with N719 based DSSC.21
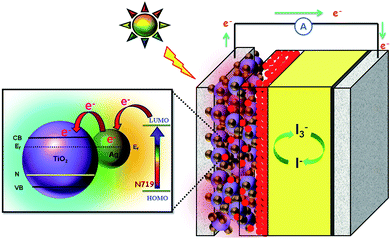 |
| | Fig. 10 Schematic functions of Ag@TiO2 nanocomposite-modified photoanode-based dye-sensitized solar cells. | |
4. Conclusion
In conclusion, plasmonic silver nanoparticles modified titania (Ag@TiO2) nanocomposite materials with various Ag contents were synthesized by simple chemical reduction method without using any stabilizer and surfactants. The as-prepared Ag@TiO2 plasmoinc nanocomposite materials were used as photoanode in the dye-sensitized solar cells to investigate the solar to electrical energy conversion ability. The incorporation of Ag on the TiO2 surface significantly influenced the optical properties in the region of 400–500 nm because of the surface plasmon resonance effect and the formation of 2–4 nm sized Ag nanoparticles on the TiO2 was confirmed through the HRTEM. The DSSC assembled with the Ag@TiO2-plasmonic photoanode demonstrated an enhanced solar-to-electrical energy conversion efficiency (4.86%) than that of bare TiO2 (2.57%) under an AM 1.5G simulated solar irradiation of 100 mW cm−2, due the surface plasmon resonance effect of Ag nanoparticles present in the nanocomposites. The influence of the Ag content on the overall efficiency was also investigated, and the optimized Ag content with TiO2 was found to be 2.5 wt%. The enhanced solar energy conversion efficiency of the Ag@TiO2 plasmonic nanocomposite makes it a promising alternative to conventional photoanode-based DSSCs.
Acknowledgements
The authors wish to express their gratitude to the Ministry of Higher Education and the University of Malaya for sanctioning a High Impact Research Grant (UM.C/625/1/HIR/MOHE/SC/21) and UMRG Programme Grant (RP007C/13AFR). The authors also wish to thank the Synchrotron Light Research Institute, Nakhon Ratchasima, Thailand for the XPS measurements.
References
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef.
- Q. Tao, X. Zhao, Y. Chen, J. Li, Q. Li, Y. Ma, J. Li, T. Cui, P. Zhu and X. Wang, RSC Adv., 2013, 3, 18317–18322 RSC.
- H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Mater. Chem., 2012, 22, 15475–15489 RSC.
- J. van de Lagemaat, N. G. Park and A. J. Frank, J. Phys. Chem. B, 2000, 104, 2044–2052 CrossRef CAS.
- N. Kopidakis, N. R. Neale, K. Zhu, J. van de Lagemaat and A. J. Frank, Appl. Phys. Lett., 2005, 87, 202106 CrossRef PubMed.
- Y. Lai, H. Zhuang, K. Xie, D. Gong, Y. Tang, C. L. L. Sun and Z. Chen, New J. Chem., 2010, 1335–1340 RSC.
- J. M. Macak, F. Schmidt-Stein and P. Schmuki, Electrochem. Commun., 2007, 9, 1783–1787 CrossRef CAS PubMed.
- L. Yang, D. He, Q. Cai and C. A. Grimes, J. Phys. Chem. C, 2007, 111, 8214–8217 CAS.
- H. Zhao, Y. Chen, X. Quan and X. Ruan, Chin. Sci. Bull., 2007, 52, 1456–1461 CrossRef CAS PubMed.
- D. M. Schaadt, B. Feng and E. T. Yu, Appl. Phys. Lett., 2005, 86, 063106 CrossRef PubMed.
- C. F. Eagen, Appl. Opt., 1981, 20, 3035–3042 CrossRef CAS PubMed.
- J. J. Mock, M. Barbic, D. R. Smith, D. A. Schultz and S. Schultz, J. Chem. Phys., 2002, 116, 6755–6759 CrossRef CAS PubMed.
- G. Zhao, H. Kozuka and T. Yoko, Sol. Energy Mater. Sol. Cells, 1997, 46, 219–231 CrossRef CAS.
- K.-C. Lee, S.-J. Lin, C.-H. Lin, C.-S. Tsai and Y.-J. Lu, Surf. Coat. Technol., 2008, 202, 5339–5342 CrossRef CAS PubMed.
- C. Wen, K. Ishikawa, M. Kishima and K. Yamada, Sol. Energy Mater. Sol. Cells, 2000, 61, 339–351 CrossRef CAS.
- Y. Binyu, L. Kar Man, G. Qiuquan, L. Woon Ming and Y. Jun, Nanotechnology, 2011, 22, 115603 CrossRef.
- C.-S. Chou, R.-Y. Yang, C.-K. Yeh and Y.-J. Lin, Powder Technol., 2009, 194, 95–105 CrossRef CAS PubMed.
- J. Qi, X. Dang, P. T. Hammond and A. M. Belcher, ACS Nano, 2011, 5, 7108–7116 CrossRef CAS PubMed.
- C. Photiphitak, P. Rakkwamsuk, P. Muthitamongkol, C. Sae-Kung and C. Thanachayanont, Int. J. Photoenergy, 2011, 2011, 357979 CrossRef.
- S. Chen, J. Li, K. Qian, W. Xu, Y. Lu, W. Huang and S. Yu, Nano Res., 2010, 3, 244–255 CrossRef CAS.
- Z. Tian, L. Wang, L. Jia, Q. Li, Q. Song, S. Su and H. Yang, RSC Adv., 2013, 3, 6369–6376 RSC.
- A. Pandikumar, K. Sivaranjani, C. S. Gopinath and R. Ramaraj, RSC Adv., 2013, 3, 13390–13398 RSC.
- S. P. Lim, N. M. Huang, H. N. Lim and M. Mazhar, Int. J. Photoenergy, 2014, 2014, 12 CrossRef PubMed.
- J. Du, J. Zhang, Z. Liu, B. Han, T. Jiang and Y. Huang, Langmuir, 2006, 22, 1307–1312 CrossRef CAS PubMed.
- N. Chandrasekharan and P. V. Kamat, J. Phys. Chem. B, 2000, 104, 10851–10857 CrossRef CAS.
- G. L. Chiarello, M. H. Aguirre and E. Selli, J. Catal., 2010, 273, 182–190 CrossRef CAS PubMed.
- K. Sivaranjani and C. S. Gopinath, J. Mater. Chem., 2011, 21, 2639–2647 RSC.
- C. Su, L. Liu, M. Zhang, Y. Zhang and C. Shao, CrystEngComm, 2012, 14, 3989–3999 RSC.
- D. Briggs, Surf. Interface Anal., 1981, 3, 1 CrossRef.
- L. Wang, L. Jia and Q. Li, Mater. Lett., 2014, 123, 83–86 CrossRef CAS PubMed.
- K. Guo, M. Li, X. Fang, X. Liu, B. Sebo, Y. Zhu, Z. Hu and X. Zhao, J. Power Sources, 2013, 230, 155–160 CrossRef CAS PubMed.
- S. G. Kim, M. J. Ju, I. T. Choi, W. S. Choi, H.-J. Choi, J.-B. Baek and H. K. Kim, RSC Adv., 2013, 3, 16380–16386 RSC.
- P. S. Archana, A. Gupta, M. M. Yusoff and R. Jose, Phys. Chem. Chem. Phys., 2014, 16, 7448–7454 RSC.
- J. Nissfolk, K. Fredin, A. Hagfeldt and G. Boschloo, J. Phys. Chem. B, 2006, 110, 17715–17718 CrossRef CAS PubMed.
- Q. Wang, Z. Zhang, S. M. Zakeeruddin and M. Grätzel, J. Phys. Chem. C, 2008, 112, 7084–7092 CAS.
- S. Lin, K. Lee, J. Wu and J. Wu, Sol. Energy, 2012, 86, 2600–2605 CrossRef CAS PubMed.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS PubMed.
- P. Christopher, D. B. Ingram and S. Linic, J. Phys. Chem. C, 2010, 114, 9173–9177 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra05689b |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a