
Highly-crystalline γ-MnS nanosaws†
Abstract
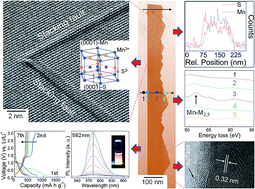
We report here a straightforward method to fabricate single-crystal wurtzite-structured manganese sulfide nanosaws (γ-MnS NSs) via a chemical vapor deposition technique, which can be extended to different types of binary chalcogenide-based 1D building blocks. Advanced spectroscopy and electron microscopy techniques were applied to reveal the structure, morphology and composition of these nanostructures. Our studies indicate that γ-MnS NSs are of high crystalline quality and purity, exhibit high aspect ratio and have extrusive teeth with average tip size of approximately a couple of atoms. These NSs are structurally stable and do not present divergence in surface energy. The formation of the backsaw is governed by a vapor–liquid–solid growth mechanism, whereas the teeth are a result of a self-catalyzed growth process induced by the Mn-terminated (0001) surface. Our optical and electrochemical analyses indicate that γ-MnS NSs can be used as efficient visible light emitters with an activation energy of ∼40 meV, and as anode materials with improved cycling stability for LIBs. This bottom-up approach represents a step ahead to provide promising and potential Mn-based materials for high-performance LIB technology and optoelectronics, and adds a new member to the family of saw-like wurtzite-structured nanoribbons, such as CdSe and ZnS.
 Please wait while we load your content...
Please wait while we load your content...

