
Thickness-dependent structural and transport behaviors in the platinum–Nafion interface: a molecular dynamics investigation
Xiao-yong Zhang and
Yi-hong Ding*
State Key Laboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Changchun 130023, People's Republic of China. E-mail: yhdd@jlu.edu.cn
First published on 5th September 2014
Abstract
Structures and transport behaviors around the ionomer–catalyst interface in polymer electrolyte membrane fuel cells (PEMFCs) have aroused great research interests in recent years. Herein, classical molecular dynamics simulation method is used to investigate the interfacial self-assembly phenomena of three fully hydrated (λ = 23) Nafion films with thicknesses of 2.4, 5.0 and 7.3 nm on the platinum surface. Interestingly, it is found that in the vicinity of the platinum surface, there is an ultra-dense adhesive ionomer layer with a thickness of 0.5 nm, whose compositions are not affected by the hydration levels and film thickness. Due to the lack of sulfonate groups, the Nafion ionomer in regions away from the Pt slab are reorganized in different patterns for films with different thicknesses. Besides this, we have found a thickness-dependence of the wetability of the surfaces exposed to the air in these fully hydrated films. It is also shown that the transport properties of hydronium ions and water molecules in the interfacial films are closely related to film morphologies. Water molecules in the 5.0 nm film are found to possess the lowest mobility as a result of the weakest connectivity of the hydrophilic channels, while in the 7.3 nm film, water diffusion is the fastest since the water channels are most ideally connected throughout this film. Notably, though water molecules cannot be retained inside the ultrathin 2.4 nm film, they could mostly develop into linear hydrophilic channels over the ionomer matrix, which can also provide transport pathways for hydrophilic species without interruption.
1. Introduction
Currently, there are increasing research activities in polymer electrolyte membrane fuel cells (PEMFCs) due to their promising applications in diverse fields, such as automobiles, portable electronics, and stationary power generators.1 PEMFCs have the advantages of high energy density, zero emissions, and high theoretical efficiency, which can make PEMFCs valuable alternatives to conventional fossil fuels. However, further efforts in reducing the cost and improving the performances are still needed to make their large-scale commercialization possible.The heart of a typical fuel cell is the membrane electrode assembly (MEA), in which a proton exchange membrane (PEM), such as Nafion, is coated with anode and cathode catalyst layers (CLs). Particularly, the three-phase boundary zone in the CLs consisting of catalyst, reactants and electrolyte is the place where the electrochemical reactions, ion conduction and multiphase fluid flow take place. During the process of fabricating CLs, platinum nanoparticles are randomly dispersed onto the porous supporting materials (carbon black or graphitized carbon) to develop agglomerates of dimensions in the scale of 100 nm. Meanwhile, Nafion ionomers would impregnate available pore spaces and coat the agglomerates of Pt/C particles. This process generally results in a porous network in CLs, which is critical for the performance of fuel cell. Lots of researches have focused on studies of CL morphology and composition, local distribution of reactants and reaction rate in them and aspects of global CL performance.2
Given the fact that the ionomer films covering the carbon agglomerates are only of a few nanometers,3 one could expect the drastically different structural and transport behaviors for polymers when confined to a thin film compared to the bulk Nafion membrane due to the differences in microstructure of the two materials.4 Although lots of experimental and computational studies have been devoted to probing the macroscopic and microscopic structures of bulk Nafion membrane,5 limited information was known about the phase behaviors of the ultrathin films coated on the Pt/C particles in CLs. This is largely due to the difficulties to probe the high dynamic and opaque heterogeneous CL nanostructures using current experimental techniques.6 Instead of using real catalysts layers of fuel cells, the researchers adopted a flat electrode to measure the Pt–Nafion interface. By means of this technique, several interfacial chemical groups, e.g. hydrated protons, water molecules, sulfonate anions and ether groups in the Nafion side chain, have been identified by means of voltammetric approach7 and spectroscopic techniques, including IR spectroscopy,8 surface-enhanced Raman spectroscopy,9 etc. There also have been several reports studying on the effects of the relative humidity level,10 Pt loading11 and film thickness12 on the interfacial structures and gaseous transport behaviors. Ohira et al. observed an abrupt decrease in proton conduction for ∼5 nm ionomer film at the interface with platinum.12b In their report, the reason was ascribed to the reduction in the number of active proton conductive pathways and/or the connectivity of the proton path network with film thickness decreasing. Most recently, Weber et al. conducted a thorough study of the behavior of Nafion thin films on carbon, gold and platinum substrates.12a Their results revealed that for all substrates, as the films' thickness is decreased, there is an initial decrease in swelling followed by a subsequent increase for film thicknesses below ≈20 nm due to a disordering of the film hydrophilic/hydrophobic structure.
Aside from those experimental reports, several classical molecular dynamic (MD) simulations have also helped better understandings of the morphology and transport dynamics of catalyst layer within a PEMFC.13–17 Keffer and coworkers studied the electrode/electrolyte interface by analyzing the effect of hydration levels, surface wetting ability, film thickness, carbon support type (hydroxylated, epoxidized and pristine) and the presence of a catalyst (Pt or PtO) on the interfacial structure or the transport property in the CLs.15 Djilali et al. also discussed the effect of Pt nanoparticles size on the microstructure of catalyst layer.16 Their results indicated that water molecules prefer to be oriented away from the graphite surface, and to be specially attracted by Pt atoms and side chains of Nafion. In a recent publication, Malek et al. introduced a mean-field like model to describe the interactions between ionomer and substrate.17 In their work, they addressed the significant modifications of the global wetting properties of the unstructured substrate surfaces on proton transport and film conformations.
It has been well known that as the film becomes thinner, the interactions of substrate/Nafion as well as confinement effect become increasingly important in affecting the microstructure of Nafion film.4,12 Strikingly, we noted that it is still not well understood about what would happen to the microphase separation and transport properties in Nafion ultrathin (less than 10 nm) films at the interface with Pt from a molecular level. In the present work, we conducted a molecular dynamics simulation about the self-assembly phenomena of three ultrathin films on the platinum surface to disclose the effect of confinement on interfacial morphologies and transport behaviors. The effect of hydration levels on the structural and transport properties was discussed as well.
2. Simulation method
2.1 Model construction
In the present work, an infinite slab is chosen to model the platinum electrode without considering its geometric effects, such as size, shape, the ratio of surface area to volume and surface index facets. In a simulation box with dimensions of 6 nm × 6 nm × 50 nm, 3364 platinum atoms in seven layers, whose X/Y direction size was rightly accommodated with the simulation box, were initially located at the bottom of the simulation box. The mixture of Nafion, water molecules and hydronium ions were randomly packed in regions right above the Pt slab along Z-axis using Packmol package.18 Taking the 10 oligomers case as an example, the mixture would be confined in a cuboid next to the slab with length 6 nm (along X, same as the X length of the simulation cell), width 6 nm (along Y, same as the Y length of the simulation cell) and height 7 nm (along Z, perpendicular to the Pt the slab) and molecules were randomly distributed with every pair of atoms of different molecules separated by at least 2.0 Å. Then, sufficient space was left along the Z direction to avoid interactions between ionomers with platinum of the neighboring image.The Nafion oligomer employed in our work consists of ten monomers with an equivalent weight of about 1100 g, as illustrated in Fig. 1, which is shown as ten hydrophilic SO3− terminated side chains evenly spaced by seven hydrophobic –CF2–CF2– backbone groups. The sulfonic acid groups in Nafion ionomer were assumed to be completely deprotonated and ionized. In order to maintain charge neutrality of the system, hydronium ions were added, the numbers of which were equal to the total number of solfonate groups in the corresponding system. To investigate the effect of ionomer thickness, we considered three individual interfacial systems with film thickness of 2.4, 5.0 and 7.3 nm at fully hydration level (λ = 23), which respectively contained 5, 10 and 15 oligomers. Each system consists of four simulations with different water contents of λ = 1, 5, 13 and 23, where λ denotes the total number of water molecules per sulfonic acid site.
 | ||
| Fig. 1 Chemical structure of Nafion monomer. The circles stand for the head or tail atoms in the repeat unit to form oligomers. | ||
2.2 Force field
The OPLS all-atom force field in GROMACS 4.5.5 software19 was employed in this work to unravel the microscopic structure of hydrated Nafion thin film on the platinum slab. The potential parameters of bond stretching, bending, dihedral torsion, and intramolecular and intermolecular non-bonded interactions via the 12-6 Lennard-Jones (LJ) potential and coulombic interactions in Nafion ionomers were taken from the work of Lu et al.20 To compute the non-bonded interactions, only atom pairs separated by at least three bonds were considered. Lorentz-Berthelot mixing rules were incorporated in this potential form to describe the interactions between different types of atoms. The model for hydronium ions was similar to that of Goddard et al.21 Extended simple point charge model (SPC/E) for water molecules with a flexible OH bond was used in order to produce a reasonable density and diffusion constant.22To model the interaction between the Pt atoms and Nafion polymers, it is very important to choose a suitable force field (or potential). The LJ potentials have been extensively applied to study various large systems (∼106 atoms) containing neutral atoms and molecules, especially the noble gas atoms.23 Yet, traditional LJ potentials usually cause very huge deviation when applied to metals.24 Promisingly, it was recently pointed out that the performance of LJ potential with appropriate parameters for several face-centered cubic metals (Ag, Al, Au, Cu, Pt, etc.) is comparable to tight-binding and embedded atom models at up to million times lower computational cost.25a Such a refined LJ potential for Pt has been successfully adopted to describe the interactions of Pt–peptide25b and Pt–amino acids.25c In this work, to describe the interactions between Nafion and platinum, we adopted the 12-6 LJ potential for uncharged platinum atom with the same parameters used in ref. 25a. In all simulations, periodic boundary conditions (PBC) were applied in X, Y and Z directions and platinum atoms were held fixed at their initial position.
2.3 Simulation details
In total, 12 cases of simulations were conducted. The initial configurations were firstly relaxed to the local energy minima using steepest descent optimization method. In order to obtain a reasonable equilibrium structure as fast as possible, we adopted an annealing procedure, including a long enough NVT simulation at 600 K until the energies of the system kept constant, followed by cooling from 600 K to 300 K in 5 ns, 5 ns NVT simulation at 300 K and heating from 300 K to 600 K in 5 ns with the temperature reassigned every 0.1 ps. After repeating the annealing process three times, the resulting configurations were quenched to 300 K as an initial structure for the NVT production run at 300 K for 40 ns. For each simulation, the last 10 ns of the trajectory at 50 ps intervals were saved to analysis the interfacial structures and to collect trajectories of water and hydronium ions. At the beginning of the simulation, we switched off the electrostatic interactions and gradually restored the LJ parameters of those atoms except platinum from zero to the normal value in a period of 10 ns. After that, the annealing procedure was conducted with the electrostatic interactions on. Normally, the memory for the initial structures would be lost after such a process. This simulation scheme has been used previously to get reasonable simulation results for interfacial systems consisting of metal and polymers.15fModified Berendsen thermostat was used to control the temperature, and particle mesh Ewald algorithm (PME) was applied to calculate the long-range electrostatics interactions with a cut-off distance of 1.0 nm and interpolation order of 4. The leap-frog method implemented in Gromacs package was adopted for solving and integrating Newton's equations of motion. In molecular dynamics simulation, the selection of time step is very important to determine the intramolecular motion. Usually, 1 fs time step is used for considering several logical reasons.26 One of them is that stable dynamics will be executed only if we use a relatively smaller time step than the period of the highest vibrational frequency in the molecule. However, a much smaller step size (much less than 1 fs) in turn limits the ability to fully sample the phase space. Besides, if we choose a much larger time step, the system would become uncontrolled due to the extremely large changes in velocity and position when going from one step to the next step. In the present work, to balance the calculation resources and simulation stability, a time step of 1 fs was considered in the present work.
The long-rang mobility of hydronium ions and water molecules are estimated from the slope of the mean square displacement (MSD) using Einstein relationship, with expression showing as follows:27
 | (1) |
In order to examine the availability of the current force fields for Nafion, hydronium ions and water molecules, we have also simulated the bulk Nafion solution including 4 Nafion oligomers and 40 hydronium ions at 300 K. The water content was set to λ = 1, 5, 13 and 23. An additional NPT production run of 40 ns at 1 atm were performed to attain the final density after the annealing strategy as was described for the calculation of the Pt–Nafion interfacial systems. The simulated system densities and self diffusion coefficients of hydronium ions (DH) and water molecules (DW) at different water contents were listed in Table 1. Clearly, these values are in reasonable agreement with previous reports,28 implying that the current force fields can suitably describe the mixture of hydronium ions, water molecules and Nafion ionomers.
| λ | ρsim (g cm−3) | DH (10−5 cm2 s−1) | DW (10−5 cm2 s−1) |
|---|---|---|---|
| 1 | 1.950 | 0.006 | — |
| 5 | 1.859 | 0.013 | 0.040 |
| 13 | 1.728 | 0.108 | 0.407 |
| 23 | 1.596 | 0.180 | 0.880 |
3. Results and discussion
3.1 Morphology of ultrathin films
Distributions of fluorine atoms (F) in Nafion CF2 backbone, sulfur atoms (S) in Nafion pendant side chains, oxygen atoms (OW) in water molecules and oxygen atoms (OH) in hydronium ions along the Z direction for systems with different amounts of Nafion oligomers are displayed in Fig. 2. Based on the distribution of CF2 and solfonate groups, the relationship between film thickness and ionomer contents at various hydration levels is shown in Table 2. The thickness of a film is mainly affected by two factors, i.e., the Nafion contents and the hydration levels. Upon increasing the ionomer contents, hydration level becomes more pronounced in altering the film thickness. In the system with the smallest amounts of ionomer (5 oligomers), the film thickness only slightly increases from 2.0 nm to 2.4 nm when increasing the hydration level from 1 to 23. However, nearly 2.1 nm increment in thickness is expected for the system with 15 oligomers in the same situation. This can be explained by the distribution of water molecules upon hydration. In the case of 5 oligomers, as shown in Fig. 2a, most of the ionomers are tightly adsorbed on the Pt slab. This would lead most of the water molecules to distribute over the external surface of the film, which would have little contribution to the thickness of film. In the cases of higher oligomer contents (10 oligomers or 15 oligomers), water molecules are retained inside the films, and then the films can be swollen with larger increment in thickness with the increase of water contents.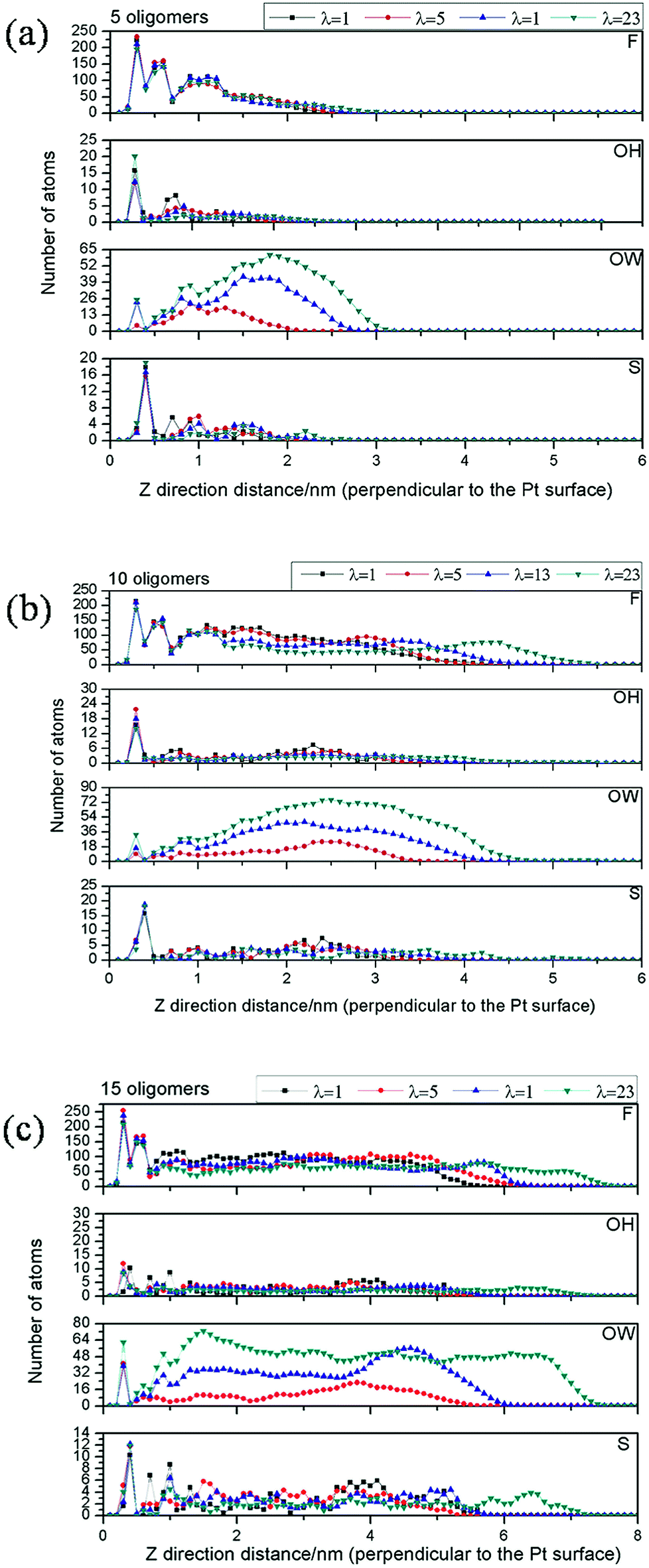 | ||
| Fig. 2 Number density distributions of fluorine atoms (F) in Nafion CF2 backbone and sulfur atoms (S) in Nafion pendant side chains, oxygen atoms in water (OW) and hydronium ions (OH) along the direction perpendicular to the platinum surface for the systems with (a) 5 oligomers, (b) 10 oligomers and (c) 15 oligomers. The location of Pt surface is positioned at zero point in the X axis. | ||
| Water content λ | 5 oligomers (nm) | 10 oligomers (nm) | 15 oligomers (nm) |
|---|---|---|---|
| 1 | 2.0 | 3.5 | 5.2 |
| 5 | 2.2 | 3.6 | 5.8 |
| 13 | 2.3 | 4.2 | 6.0 |
| 23 | 2.4 | 5.0 | 7.3 |
As shown in Fig. 2, the atomic distributions in the interfacial films are found to present density layering order along the Z direction. Significantly, there exists a compact adhesive ionomer layer with thickness of 0.5 nm above the Pt slab for all the interfacial systems. This may be due to significant nanoconfinement interactions of Pt towards Nafion ionomer. Interestingly, the distribution peak of sulfonate groups in the compact layer is much higher than that in the other regions away from the platinum slab. In addition, the numbers of solfonate groups contributing to this peak seem to be little influenced by the hydration levels and Nafion contents, which can be attributed to the stronger interactions between sulfonic sites and Pt atoms. Upon increasing the Nafion content, the distribution of CF2 groups is much more influenced by the hydration levels. In regions away from the platinum, the films can undergo significant reorganization of the hydrophobic/hydrophilic domains in contrast to the nearly no change of the ionomer compositions in the vicinity of the platinum surface.
For the three systems with different amounts of oligomers, the final snapshots of their self-organized morphologies at λ = 23, where they are fully hydrated, are displayed in Fig. 3. The hydrophobic/hydrophilic separation situations in the three systems are rather different. For the system with the smallest amounts of ionomers (2.4 nm) in Fig. 3a, most of the water molecules cluster over the external surface of the film and consequently linear hydrophilic channels develop along the Y direction. Differently, the water molecules are mainly buried inside the films with the medium (5.0 nm) and the highest (7.3 nm) ionomer contents. In the former case, the hydrophilic domains (mixtures of water molecules, hydronium ions and Nafion side chains) tend to be isolated from each other by the hydrophobic domains. This may be due to the fact that large amounts of the sulfonate groups have been contributed to covering the Pt surface so that there would lack hydrophilic groups (e.g., solfonate sites) to interconnect these hydrophilic domains. In addition, according to the distribution curves in Fig. 2b and snapshots of the film morphology in Fig. 3b, the external surface of this film is mainly covered with hydrophobic CF2 groups. At the highest ionomer content in Fig. 3c, water molecules can form highly connected hydrophilic channels both along the lateral and vertical directions. In this film, water molecules and hydronium ions are entering and passing through the Nafion phase. Around the ionomer/air interface, it is clearly shown that the hydrophilic domains (side chain and water molecules) are more pronounced than the hydrophobic domains (CF2 group).
 | ||
| Fig. 3 Morphologies of the ultrathin films deposited on the platinum slab at the water content of λ = 23 in the presence of (a) 5 oligomers, (b) 10 oligomers, and (c) 15 oligomers. Gray: CF2 backbone. Orange: side chains. Green: water. Purple: hydronium ions. Cyan: platinum slab. For each case, top (left) and side (right) views are shown. | ||
Fig. 4 shows the top views of the final snapshots of the interfacial films with 15 oligomers at λ = 5, 13 and 23, respectively. Clearly, higher water contents could guarantee better connectivities of the hydrophilic channels through the films. This is also true for the interfacial films with 5 or 10 oligomers. At λ = 5, there are large amounts of disconnected and small water clusters in the film. At a higher water content of λ = 13, water molecules gradually form larger islands. However, there are still some disconnected clusters. In the case of λ = 23 in Fig. 4c, water molecules are so rich that highly interconnected water-flooded channels are formed through the film, which could provide continuous transport pathways for hydrophilic species.
 | ||
| Fig. 4 Top views of the final snapshots of the interfacial films with 15 oligomers at λ = (a) 5, (b) 13 and (c) 23 from left to right. The coloring mode is the same as that in Fig. 3. | ||
3.2 Interfacial transport property
It should be stressed here that since a nondissociated harmonic bond model is used for the O–H bond in water molecules and hydronium ions, the proton hopping mechanism between water molecules and hydronium ions (i.e., Grotthus mechanism or structural mechanism) is not allowed in our molecular dynamics simulation. Instead, this mechanism can be studied using ab initio simulations or self-consistent multistate empirical valence bond (SCI-MS-EVB) method.29 Clearly, there would exist qualitative differences between the simulated diffusion coefficients of hydronium ions and experimental ones, especially at high hydration levels. In this work, we mainly concentrate on the transport of water molecules through these confined films. It is known that the mobility of water molecules at interfaces can affect the overall proton conductivity and chemical reactions in CLs.6 The transport behavior of hydronium ions and proton is highly influenced by the connectivity of hydrophilic domains.29a The proceeding discussion on the simulated morphology of hydrophilic channels should provide a useful understanding of the proton conduction behavior. In the two fully hydrated films (2.4 and 7.3 nm), the former one has linear hydrophilic channels and the latter present continuous hydrophilic channels through the film. Thus, proton transport pathways could develop without interruption in both fully hydrated films. However, the transport pathways of water molecules and hydronium ions, as well as proton would be interrupted in the 5.0 nm film as a result of the poor connectivity of the hydrophilic channels. These morphology characteristics could well illustrate the experimental observation that an abrupt decrease in proton conduction for ∼5 nm ionomer film takes place at the interface with platinum.12bFig. 5a and 6a show the MSD plots of OH (oxygen atom in hydronium ions) and OW (oxygen atom in water molecules) in the bulk Nafion solution and the three interfacial films at fully hydration level (λ = 23). The log(MSD)–log(time) behavior of the mean square displacement curves and the adjusted R square values for lines of the best fit of these graphs using linear regression are shown in Fig. 5b (OH) and Fig. 6b (OW). The MSD values in all cases are well linearly proportional to the observation time and thus can be used to yield reasonable diffusion coefficients. The calculated X/Y/Z and average self diffusion coefficients of hydronium ions and water molecules at λ = 23 are provide in Table 3.
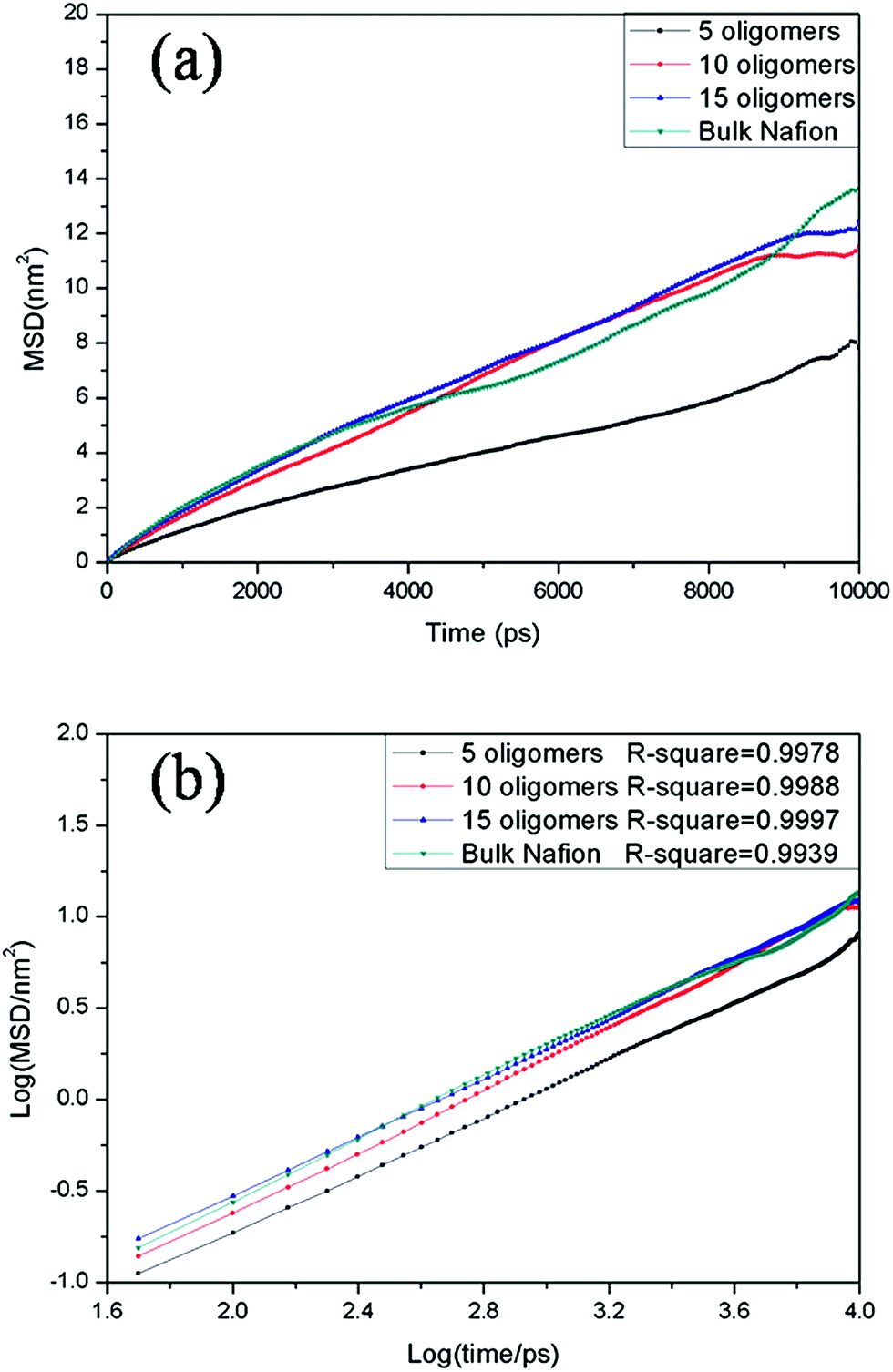 | ||
| Fig. 5 (a) Mean square displacement (MSD) curves and (b) log–log plot of mean square displacement with respect to the observation time for oxygen atom (OH) in hydronium ions at λ = 23 in films with different Nafion contents. | ||
 | ||
| Fig. 6 (a) Mean square displacement (MSD) curves and (b) log–log plot of mean square displacement with respect to the observation time of oxygen atom (OW) in water molecules at λ = 23 for films with different Nafion contents. | ||
| System | DH (10−5 cm2 s−1) | DW (10−5 cm2 s−1) | |||
|---|---|---|---|---|---|
| Average | X/Y/Z | Average | X/Y/Z | ||
| 5 oligomers | X | 0.112 | 0.167 | 0.813 | 0.013 |
| Y | 0.217 | 1.597 | |||
| Z | 0.013 | 0.828 | |||
| 10 oligomers | X | 0.214 | 0.333 | 0.658 | 0.938 |
| Y | 0.217 | 0.719 | |||
| Z | 0.091 | 0.317 | |||
| 15 oligomers | X | 0.193 | 0.226 | 1.019 | 0.921 |
| Y | 0.231 | 0.847 | |||
| Z | 0.124 | 1.289 | |||
| Bulk Nafion | 0.180 | 0.880 | |||
Clearly, the vehicular mobility of hydronium ions is much lower than that of water molecules in interfacial films. This can be explained by the uniform strong electrostatic attraction between hydronium ions and the sulfonate groups. In systems with 5 and 10 oligomers, the diffusion coefficients of hydronium ions in the Z direction are observed to be much smaller than the corresponding values in the other two directions. In the case of 5 oligomers, the ionomer film (2.4 nm) presents compact adhesive morphology with linear water channels distributed over the free surface along Y direction, which would favor the planar transport rather than the vertical transport. Moreover, this structural pattern has also affected the transport of water, i.e., water molecules mainly diffuse along the Y direction with a coefficient of 1.597 × 10−5 cm2 s−1. However, in the film (5.0 nm) with 10 oligomers, the vertical transport of hydronium ions and water molecules is blocked as most of these hydrophilic species are confined within the film. The mobility of water molecules in this film is the lowest (DW = 0.658 × 10−5 cm2 s−1) among the three interfacial films, which can be explained by the poorly connected of the hydrophilic channels, as shown in Fig. 3b. For the film (7.3 nm) with 15 oligomers, due to the forming of continuous and interconnected hydrophilic channels, water molecules can uniformly transport in all directions, and thus has the highest mobility (DW = 1.019 × 10−5 cm2 s−1).
Generally, CLs are complex heterogeneous media, which involves a hierarchy of scales. The thickness of ionomer covered on Pt/C does not appear to be homogeneous in CLs.30 In addition, the actual situations inside CLs are highly dynamics under fuel cell operating conditions. Thus, the overall transport picture might not be as simple as the present MD simulation illustrates. For example, it has been reported that the electric field could alter the aggregation conditions of hydrophilic channels in bulk Nafion solutions, and then promote the diffusion of charged ions.31 Similarly, it can be expected that with appropriate controls of the electric field, the hydrophilic channels in interfacial films could also be reorganized in an ordered way to facilitate the transport of hydrophilic species. Given a certain voltage, variation of film thickness would result in different electric gradient across the interfacial film, which may have significant effects on the interfacial morphology and transport property (e.g. diffusivity of hydrophilic species and conductivity of ions). Thus, it would be of great interest to consider the electric field in further interfacial model development.
4. Conclusion
In this work, the microscopic structures and transport behaviors of hydronium ions and water molecules at platinum–Nafion interfaces are investigated by classical molecular dynamics simulations. There are several key structural features during increasing the thickness of fully hydrated film from 2.4 nm to 7.3 nm: (1) a compact binding layer of ionomers with thickness of about 0.5 nm is observed in the vicinity of the platinum surface, and the compositions of this layer seem to be independent of the film thickness and hydration levels, (2) significant amounts of sulfonate groups in Nafion are attracted by the platinum surface, which could cause significant reorganization of the ionomers in regions away from the platinum surfaces, (3) the wetability of the external surfaces exposed to the air transforms from a linear water channel (2.4 nm film), to a hydrophobic surface (5.0 nm film) with most of water molecules deeply inside the film, and finally to a surface both including hydrophobic and hydrophilic sites (7.3 nm film). Due to the weakest connectivity of the hydrophilic channels, the lowest mobility of water molecules would occur in the 5.0 nm film, while the 7.3 nm film holds the highest water diffusion ability, in agreement with the ideal connectivity of the hydrophilic channels in this film. These results can well interpret the recent experimental observation that due to the difference of microstructures affected by film thickness, an abrupt decrease in proton conduction for ∼5 nm ionomer film takes place at the interface with platinum.12b Although it is impossible to retain water molecules inside the 2.4 nm film, the linear and continuous hydrophilic channels over the ionomer matrix can still provide transport pathways for hydronium ions and water molecules. Due to the experimental difficulties to probe the internal morphologies and transport properties in ultrathin films, we believe that our simulation work could provide valuable molecular-level information to understand the thickness-dependent interfacial structures and transport behaviors in CLs.Acknowledgements
This work was funded by the National Basic Research Program of China (973 Program) (2012CB932800), Development Program of China under Grant no. 2010CB327701, the National Natural Science Foundation of China (no. 21273093, 20773054, 21073074). The reviewers' comments and suggestions are greatly appreciated.References
- (a) M. Z. F. Kamarudin, S. K. Kamarudin, M. S. Masdar and W. R. W. Daud, Int. J. Hydrogen Energy, 2013, 38, 9438–9453 CrossRef CAS PubMed; (b) X. Z. Yuan, H. Li, S. S. Zhang, J. Martin and H. J. Wang, J. Power Sources, 2011, 196, 9107–9116 CrossRef CAS PubMed; (c) Y. Wang, K. S. Chen, J. Mishler, S. C. Cho and X. C. Adroher, Appl. Energy, 2011, 88, 981–1007 CrossRef CAS PubMed; (d) K. E. Martin, J. P. Kopasz and K. W. McMurphy, in Fuel Cell Chemistry and Operation, American Chemical Society, 2010, vol. 1040, pp. 1–13 Search PubMed; (e) N. W. Deluca and Y. A. Elabd, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 2201–2225 CrossRef CAS.
- (a) R. A. Silva, T. Hashimoto, G. E. Thompson and C. M. Rangel, Int. J. Hydrogen Energy, 2012, 37, 7299–7308 CrossRef CAS PubMed; (b) R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon and D. Wood, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed; (c) K. Malek, M. Eikerling, Q. P. Wang, T. Navessin and Z. S. Liu, J. Phys. Chem. C, 2007, 111, 13627–13634 CrossRef CAS; (d) Q. P. Wang, M. Eikerling, D. T. Song and Z. S. Liu, J. Electroanal. Chem., 2004, 573, 61–69 CrossRef CAS.
- (a) T. Soboleva, X. S. Zhao, K. Malek, Z. Xie, T. Navessin and S. Holdcroft, ACS Appl. Mater. Interfaces, 2010, 2, 375–384 CrossRef CAS PubMed; (b) Z. Xie, T. Navessin, K. Shi, R. Chow, Q. P. Wang, D. T. Song, B. Andreaus, M. Eikerling, Z. S. Liu and S. Holdcroft, J. Electrochem. Soc., 2005, 152, A1171–A1179 CrossRef CAS PubMed.
- (a) S. Kim, J. A. Dura, K. A. Page, B. W. Rowe, K. G. Yager, H.-J. Lee and C. L. Soles, Macromolecules, 2013, 46, 5630–5637 CrossRef CAS; (b) M. A. Modestino, D. K. Paul, S. Dishari, S. A. Petrina, F. I. Allen, M. A. Hickner, K. Karan, R. A. Segalman and A. Z. Weber, Macromolecules, 2013, 46, 867–873 CrossRef CAS; (c) D. K. Paul, K. Karan, A. Docoslis, J. B. Giorgi and J. Pearce, Macromolecules, 2013, 46, 3461–3475 CrossRef CAS; (d) S. K. Dishari and M. A. Hickner, ACS Macro Lett., 2012, 1, 291–295 CrossRef CAS; (e) S. A. Eastman, S. Kim, K. A. Page, B. W. Rowe, S. Kang, C. L. Soles and K. G. Yager, Macromolecules, 2012, 45, 7920–7930 CrossRef CAS.
- (a) G. S. Hwang, D. Y. Parkinson, A. Kusoglu, A. A. MacDowell and A. Z. Weber, ACS Macro Lett., 2013, 2, 288–291 CrossRef CAS; (b) R. Hiesgen, I. Wehl, E. Aleksandrova, E. Roduner, A. Bauder and K. A. Friedrich, Int. J. Energy Res., 2010, 34, 1223–1238 CAS; (c) K. Schmidt-Rohr and Q. Chen, Nat. Mater., 2007, 7, 75–83 CrossRef PubMed; (d) K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4586 CrossRef CAS.
- S. Holdcroft, Chem. Mater., 2013, 26, 381–393 CrossRef.
- (a) T. Masuda, F. Sonsudin, P. R. Singh, H. Naohara and K. Uosaki, J. Phys. Chem. C, 2013, 117, 15704–15709 CrossRef CAS; (b) A. M. Gómez-Marín, A. Berná and J. M. Feliu, J. Phys. Chem. C, 2010, 114, 20130–20140 CrossRef; (c) R. Subbaraman, D. Strmcnik, V. Stamenkovic and N. M. Markovic, J. Phys. Chem. C, 2010, 114, 8414–8422 CrossRef CAS.
- (a) H. Hanawa, K. Kunimatsu, M. Watanabe and H. Uchida, J. Phys. Chem. C, 2012, 116, 21401–21406 CrossRef CAS; (b) I. Kendrick, D. Kumari, A. Yakaboski, N. Dimakis and E. S. Smotkin, J. Am. Chem. Soc., 2010, 132, 17611–17616 CrossRef CAS PubMed; (c) K. Kanamura, H. Morikawa and T. Umegaki, J. Electrochem. Soc., 2003, 150, A193–A198 CrossRef CAS PubMed; (d) Y. Ayato, K. Kunimatsu, M. Osawa and T. Okada, J. Electrochem. Soc., 2006, 153, A203–A209 CrossRef CAS PubMed.
- J. B. Zeng, D. I. Jean, C. X. Ji and S. Z. Zou, Langmuir, 2011, 28, 957–964 CrossRef PubMed.
- (a) H. Noguchi, K. Taneda, H. Naohara and K. Uosaki, Electrochem. Commun., 2013, 27, 5–8 CrossRef CAS PubMed; (b) J. Chlistunoff, F. Uribe and B. Pivovar, ECS Trans., 2006, 1, 137–146 CAS.
- (a) T. A. Greszler, D. Caulk and P. Sinha, J. Electrochem. Soc., 2012, 159, F831–F840 CrossRef CAS PubMed; (b) K. Sakai, K. Sato, T. Mashio, A. Ohma, K. Yamaguchi and K. Shinohara, ECS Trans., 2009, 25, 1193–1201 CAS.
- (a) A. Kusoglu, D. Kushner, D. K. Paul, K. Karan, M. A. Hickner and A. Z. Weber, Adv. Funct. Mater., 2014, 24, 4763–4774 CrossRef CAS; (b) A. Ohira, S. Kuroda, H. F. M. Mohamed and B. Tavernier, Phys. Chem. Chem. Phys., 2013, 15, 11494–11500 RSC; (c) K. Kudo and Y. Morimoto, ECS Trans., 2013, 50, 1487–1494 CrossRef PubMed.
- E. J. Lamas and P. B. Balbuena, Electrochim. Acta, 2006, 51, 5904–5911 CrossRef CAS PubMed.
- T. Mashio, K. Malek, M. Eikerling, A. Ohma, H. Kanesaka and K. Shinohara, J. Phys. Chem. C, 2010, 114, 13739–13745 CAS.
- (a) Q. He, N. S. Suraweera, D. C. Joy and D. J. Keffer, J. Phys. Chem. C, 2013, 117, 25305–25316 CrossRef CAS; (b) Q. He, D. C. Joy and D. J. Keffer, J. Power Sources, 2013, 241, 634–646 CrossRef CAS PubMed; (c) M. E. Selvan, Q. He, E. M. Calvo-Munoz and D. J. Keffer, J. Phys. Chem. C, 2012, 116, 12890–12899 CrossRef CAS; (d) J. Liu, M. E. Selvan, S. Cui, B. J. Edwards, D. J. Keffer and W. V. Steele, J. Phys. Chem. C, 2008, 112, 1985–1993 CrossRef CAS; (e) J. Liu, S. Cui and D. J. Keffer, Fuel Cells, 2008, 8, 422–428 CrossRef CAS; (f) M. E. Selvan, Q. He, E. M. Calvo-Munoz and D. J. Keffer, J. Phys. Chem. C, 2012, 116, 12890–12899 CrossRef CAS.
- C. Cheng, K. Malek, P. Sui and N. Djilali, Electrochim. Acta, 2010, 55, 1588–1597 CrossRef CAS PubMed.
- D. Damasceno Borges, A. A. Franco, K. Malek, G. Gebel and S. Mossa, ACS Nano, 2013, 7, 6767–6773 CrossRef CAS PubMed.
- (a) L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed; (b) J. M. Martínez and L. Martínez, J. Comput. Chem., 2003, 24, 819–825 CrossRef PubMed.
- B. Hess, C. Kutzner, D. Van DerSpoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- L. Yan, S. Zhu, X. Ji and W. Lu, J. Phys. Chem. B, 2007, 111, 6357–6363 CrossRef CAS PubMed.
- S. S. Jang, V. Molinero, T. Cagin and W. A. Goddard, J. Phys. Chem. B, 2004, 108, 3149–3157 CrossRef CAS.
- H. Berendsen, J. Grigera and T. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- D. Frenkel and B. Smit, Understanding molecular simulation: from algorithms to applications, Academic Press, San Diego, 2002 Search PubMed.
- J. Uppenbrink and D. J. Wales, J. Chem. Phys., 1992, 96, 8520–8534 CrossRef CAS PubMed.
- (a) H. Heinz, R. Vaia, B. Farmer and R. Naik, J. Phys. Chem. C, 2008, 112, 17281–17290 CrossRef CAS; (b) L. Ruan, H. Ramezani-Dakhel, C.-Y. Chiu, E. Zhu, Y. Li, H. Heinz and Y. Huang, Nano Lett., 2013, 13, 840–846 CrossRef CAS PubMed; (c) S. K. Ramakrishnan, M. Martin, T. Cloitre, L. Firlej, F. J. Cuisinier and C. Gergely, J. Chem. Inf. Model., 2013, 53, 3273–3279 CrossRef CAS PubMed.
- J.-I. Choe and B. Kim, Bull. Korean Chem. Soc., 2000, 21, 419–424 CAS.
- D. C. Rapaport, The art of molecular dynamics simulation, Cambridge University Press, 2004, vol. 1, ch. 5, pp. 121–124 Search PubMed.
- (a) D. R. Morris and X. Sun, J. Appl. Polym. Sci., 1993, 50, 1445–1452 CrossRef CAS; (b) R. Devanathan, A. Venkatnathan and M. Dupuis, J. Phys. Chem. B, 2007, 111, 8069–8079 CrossRef CAS PubMed; (c) R. Devanathan, A. Venkatnathan and M. Dupuis, J. Phys. Chem. B, 2007, 111, 13006–13013 CrossRef CAS PubMed.
- (a) R. Jorn, J. Savage and G. A. Voth, Acc. Chem. Res., 2012, 45, 2002–2010 CrossRef CAS PubMed; (b) R. Devanathan, N. Idupulapati, M. D. Baer, C. J. Mundy and M. Dupuis, J. Phys. Chem. B, 2013, 117, 16522–16529 CrossRef CAS PubMed; (c) G. A. Luduena, T. D. Kühne and D. Sebastiani, Chem. Mater., 2011, 23, 1424–1429 CrossRef CAS.
- M. Lee, M. Uchida, H. Yano, D. A. Tryk, H. Uchida and M. Watanabe, Electrochim. Acta, 2010, 55, 8504–8512 CrossRef CAS PubMed.
- (a) P. Y. Chen and C. W. Hong, Fuel Cells, 2010, 10, 17–25 CAS; (b) E. Allahyarov, P. L. Taylor and H. Löwen, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 81, 031805 CrossRef.
| This journal is © The Royal Society of Chemistry 2014 |