
Polymer supported Pd catalyzed carbonylation of aryl bromides for the synthesis of aryl esters and amides†
Sk Manirul Islam*,
Kajari Ghosh,
Anupam Singha Roy‡
and
Rostam Ali Molla‡
Department of Chemistry, University of Kalyani, Kalyani, Nadia, 741235, W.B., India. E-mail: manir65@rediffmail.com; Fax: +91-33-2582-8282; Tel: +91-33-2582-8750
First published on 15th August 2014
Abstract
A polymer-anchored palladium(II) catalyst was synthesized and characterized using various spectroscopic techniques. Its catalytic activity was evaluated for the alkoxycarbonylation and aminocarbonylation reactions. These carbonylation reactions were carried out for various substituted aryl bromides using alcohols and amines. Both the reactions were optimized by varying the bases, temperature and solvents. These experiments were carried out under high CO pressure. The catalyst was very stable and can be facilely recovered and reused six times without a significant decrease in its activity and selectivity.
Introduction
Carbonylation processes using carbon monoxide (CO) as a C1 building block have proven to be a powerful tool to construct important scaffolds in natural products, pharmaceuticals, agrochemicals, and functional materials.1,2 Over the past few decades, extensive efforts have been made to develop transition-metal catalyzed carbonylation of organic halides3–5 or organic pseudohalides6,7 for the synthesis of carboxylic acids, esters, amides, and so on.Carbonylation of aryl halides with alcohols or amines catalyzed by palladium complexes is a convergent and direct route for the synthesis of aromatic esters and amides.8 Aromatic esters are important building blocks for various pharmaceuticals and agrochemicals.9 Traditionally, these esters were synthesized via reaction of the carboxylic acid with alcohols.10 Carbonylation of the aryl halides in the presence of an alcohol is an attractive alternative method that tolerates a wide range of substrates, thus demonstrating a great advantage for the synthesis of substituted aromatic esters and its derivatives.11–13 In this regard, various palladium-based catalytic systems have been explored for alkoxycarbonylation reaction.14–18
Amides are also important chemicals that exist in pharmaceuticals, natural products, and many functional materials.19,20 Arylacetamides have gained considerable attention because of their pharmacological and biological activities.21 Some heterocyclic amides are potential CNS (central nervous system)-active compounds.22 Among the methods documented for the preparation of amides, most of them require harsh conditions (>150 °C) or multistep operations.23–27 Alternatively, transition-metal catalyzed amino carbonylation has emerged as an efficient strategy towards such types of compounds.28 Aminocarbonylation is a constructive method for the direct synthesis of aromatic amides via coupling of aryl, heteroaryl, or alkynyl halides with primary/secondary amines.9
Literature reports reveal that alkoxycarbonylation and aminocarbonylation have been well explored by using a variety of homogeneous Pd complexes using various air and moisture-sensitive N/P-containing ligands.29–35 But the recovery and recycling of the homogeneous palladium catalyst is a challenging task. To get rid of these serious issues, some heterogeneous catalysts have been reported.36 However, these methods require prolonged reaction time, exotic reaction condition and the important issue is that most of these reported methods used phosphine containing complex which is toxic in nature. On the other hand most of the alkoxy and amino carbonylations used aryl iodides as the choice of substrate, and quantitative yields were reported.37,9 Therefore, developing a simple and versatile method for the preparation of aryl esters and amides from less expensive aryl bromides catalyzed by heterogeneous palladium catalyst under phosphine free condition is still a matter of interest in organic synthesis.
Herein we report the synthesis and characterization of a newly synthesized polymer anchored palladium(II) Schiff base complex and its catalytic applications towards alkoxycarbonylation and aminocarbonylation reactions of aryl bromides under the phosphine free conditions.
Experimental
Materials and instruments
Analytical-grade reagents and freshly distilled solvents were used throughout the experiment. Chloromethylated polystyrene was supplied by Sigma-Aldrich chemicals Company, USA. Other reagents were obtained from Merck Co. The chemical analysis was done by the usual procedure. The purity of solvents and substrates were checked by gas chromatography. Carbon monoxide (99.9%) purchased from IOL Bombay and palladium acetate from Arora Matthey, India, were used as received. Liquid substrates were predistilled and dried using appropriate molecular sieves. Distillation and purification of the solvents and substrates were done by standard procedures.381H NMR spectra were recorded on a Bruker DPX-400 NMR spectrometer in pure deuterated solvents with TMS as internal standard. A Perkin-Elmer 2400C elemental analyzer was used to collect micro-analytical data (C, H and N). FTIR spectra of the samples were recorded on a Perkin-Elmer FTIR 783 spectrophotometer using KBr pellets. A Mettler Toledo TGA/SDTA 851 instrument was used for the thermogravimetric (TGA) analysis. The morphology of the functionalized polystyrene and complex was analyzed using a scanning electron microscope (SEM)(ZEISS EVO40, England) equipped with EDAX facility. The metal content in the catalyst was determined using a Varian AA240 atomic absorption spectrophotometer (AAS).
Synthesis of catalyst
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) with polymer aldehyde in methanol solvent with stirring for 24 h under reflux condition to prepare the yellow Schiff base compound. The mixture was cooled to room temperature and then filtered. The residue was washed with ethanol until the filtrate became colorless and was dried under vacuum.:05) to afford the desired product.
:1 molar ratio) with polymer aldehyde in methanol solvent with stirring for 24 h under reflux condition to prepare the yellow Schiff base compound. The mixture was cooled to room temperature and then filtered. The residue was washed with ethanol until the filtrate became colorless and was dried under vacuum.:05) to afford the desired product.Results and discussion
Characterization of the polymer anchored Pd catalyst
Due to the insolubilities of the polymer-supported metal complex in all common organic solvents, its structural investigation was limited to their physicochemical properties, chemical analysis, SEM, TGA, FTIR and UV-vis spectral data. Table 1 provides the data of elemental analysis of polymer supported ligand and the polymer supported palladium catalyst. The palladium content of the polymer-supported catalysts was estimated by atomic absorption spectrometer and it suggests 8.79 wt% Pd in the catalyst.| Compound | Colour | C% | H% | Cl% | N% | Metal% |
|---|---|---|---|---|---|---|
| PS-AMP | Yellow | 75.50 | 5.96 | 5.89 | 2.32 | — |
| PS-AMP-Pd | Brown | 68.11 | 5.34 | 4.09 | 2.37 | 8.79 |
The attachment of metal onto the support was confirmed by comparison of the FT-IR spectra (Fig. 1) of the polymers before and after loading with metals, both in the mid-IR (4000–400 cm−1) region. The IR spectrum of pure chloromethylated polystyrene has an absorption band at 1261 cm−1 due to the C–Cl group, which was weak in the ligand and in the catalyst. Aldehyde functionalized polystyrene, PS-CHO shows its characteristic peak of CO at 1702 cm−1 (ESI, Fig. S1†) which was strongly reduced in the ligand and catalyst (Fig. 1). IR spectra show a stretching vibration for –CH2 at 2919 cm−1 for the polymer bound ligand and its complex. The stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond appeared at 1698 cm−1 for the polymer-bound Schiff base ligand which is shifted to 1690 cm−1 in the metal complex.40,41 Characteristic peak of free OH appeared at 3377 cm−1 for the ligand and its intensity is reduced in the complex.42 A peak is observed at 1246 cm−1 (v C–O, phenolic) in the ligand and at 1243 cm−1 in the complex.43 The polystyrene anchored Pd(II) complex exhibited important IR peaks at 1575 cm−1 (vassym COO bridged), 1358 cm−1 (vsym COO bridged) and 1733 cm−1 (v CO),44 which suggest the most probable structure of our catalyst is depicted in Scheme 1. Peaks for Pd–N45 and Pd–O46 appeared in the far-IR region.
N bond appeared at 1698 cm−1 for the polymer-bound Schiff base ligand which is shifted to 1690 cm−1 in the metal complex.40,41 Characteristic peak of free OH appeared at 3377 cm−1 for the ligand and its intensity is reduced in the complex.42 A peak is observed at 1246 cm−1 (v C–O, phenolic) in the ligand and at 1243 cm−1 in the complex.43 The polystyrene anchored Pd(II) complex exhibited important IR peaks at 1575 cm−1 (vassym COO bridged), 1358 cm−1 (vsym COO bridged) and 1733 cm−1 (v CO),44 which suggest the most probable structure of our catalyst is depicted in Scheme 1. Peaks for Pd–N45 and Pd–O46 appeared in the far-IR region.
 | ||
| Fig. 1 FT-IR spectra of polymer anchored ligand (a) and polymer anchored Pd(II) catalyst (b). | ||
 | ||
| Scheme 1 Synthesis of polymer anchored Pd(II) complex. | ||
The electronic spectra (Fig. 2) of the polymer-supported metal complex were recorded in diffuse reflectance spectrum mode as MgCO3/BaSO4 disks. The present Pd(II) complex shows a broad spectrum with few shoulders at a region of 260–530 nm. It may exhibit three spin-allowed d–d transitions. The bands are observed at 265–285 nm, 340–380 nm and 450–500 nm, which may be designated as 1A1g → 1Eg, 1A1g → 1B1g and 1A1g → 1A2g transitions respectively.47,48
 | ||
| Fig. 2 DRS-UV vis spectra of polymer anchored Pd(II) complex. | ||
Field emission-scanning electron micrographs of single bead of polymer anchored ligand (PS-L) and its complex (PS-AMP-Pd) were recorded and it was observed that the morphological changes occurred on the polystyrene beads at various stages of the synthesis. The SEM images of polymer anchored ligand (A) and the immobilized palladium(II) complex (B) on functionalized polymer are shown in Fig. 3. The pure chloromethylated polystyrene bead has a smooth surface while polymer anchored ligand and complex show slight roughening of the top layer of polymer beads. This roughening is relatively more in complex. Also the presence of palladium metal can be further proved by energy dispersive spectroscopy analysis of X-rays (EDAX) (Fig. 4) which suggests the formation of metal complex with the polymer anchored ligand.
 | ||
| Fig. 3 FE SEM images of polymer anchored ligand (A) and polymer anchored Pd(II) complex (B). | ||
 | ||
| Fig. 4 EDX images of polymer anchored ligand (A) and polymer anchored Pd(II) complex (B). | ||
Thermal stability of complex was investigated using TGA at a heating rate of 10 °C min−1 in air over a temperature range of 30–600 °C. TGA curve of polymer anchored Pd(II) complex is shown in Fig. 5. Polymer anchored palladium(II) complex decomposed at 360–400 °C. So from the thermal stability, it concludes that polymer anchored metal complex degraded at considerably higher temperature.
 | ||
| Fig. 5 TGA curve of polymer anchored Pd(II) complex. | ||
Catalytic activities
Since supported palladium catalyst exhibit high catalytic activity in a wide range of industrially important processes and have been extensively studied for C–C coupling,49 thus we decided to investigate the catalytic activity of the PS-AMP-Pd in the field of carbonylation reactions (Scheme 2). | ||
| Scheme 2 Palladium catalyzed alkoxy and amino carbonylation of aryl bromides. | ||
Alkoxycarbonylation reaction of aryl bromide and alcohol catalyzed by PS-AMP-Pd
Alkoxycarbonylation was carried out with bromobenzene and ethyl alcohol under CO pressure and maintaining basic medium using the polymer anchored Pd(II) catalyst. Ethyl benzoate was obtained as a product with 96% yield (Scheme 3). | ||
| Scheme 3 Alkoxycarbonylation of bromobenzene with ethanol. | ||
Initially, the reaction was carried out with various inorganic bases, such as K2CO3 (80%) and Cs2CO3 (79%), and organic bases, such as DBU (85%), NaOEt (91%) and Et3N (96%). Because Et3N provides the maximum yield of the corresponding product, it was used for further study. The temperature effect for the reaction was also studied (Table 2). No profound increase in the yield of the desired product was observed when the reaction temperature was increased from 70 to 100 °C; therefore, 70 °C was considered as an optimum reaction temperature for further studies.
| Entry | Base | Temperature (°C) | Conversion (%) |
|---|---|---|---|
| a Reaction conditions: bromobenzene (1 mmol), alcohol (5 mL), PS-AMP-Pd (4 × 10−4 mmol), base (3 mmol), 1 atm CO pressure, temp. (70 °C), time (3 h). | |||
| 1 | K2CO3 | 70 | 80 |
| 2 | Cs2CO3 | 70 | 79 |
| 3 | DBU | 70 | 85 |
| 4 | NaOAc | 70 | 91 |
| 5 | Et3N | 70 | 96 |
| 6 | Et3N | 90 | 96 |
| 7 | Et3N | 100 | 96 |
The reaction was carried out at 1 atm of CO pressure, providing an excellent yield of the desired product within 3 h. Various alcohols were reacted with bromobenzene and the results are summarized in Table 3. Changing the alcohol substrate from a primary (MeOH, EtOH) (Table 3, entry 1 and 2) to a secondary alcohol (iPrOH) (Table 3, entry 3) did not have a significant effect on product yield. However, moving to a tertiary alcohol (tBuOH) was not successful, a low product yield being obtained presumably due to steric bulk (Table 3, entry 4). Excellent yield was also obtained with benzyl alcohol (Table 3, entry 5). It was observed that among all the alcohols used, EtOH gave the maximum yield with bromobenzene. Hence, the finalized reaction conditions were the following: base, Et3N; temperature, 70 °C; solvent, alcohol (also as a nucleophile, here EtOH); time, 3 h; and 1 atm of CO pressure. These reaction parameters were then successfully applied for carbonylation of a variety of aryl bromides with EtOH (Table 4, entries 1–17).





| Entry | Bromides | Product | Yieldb (%) | TON |
|---|---|---|---|---|
| a Reaction conditions: aryl bromide (1 mmol), ethanol (5 mL), PS-AMP-Pd (4 × 10−4 mmol), Et3N (3 mmol), CO (1 atm), temp (70 °C), time (3 h).b Isolated yield. | ||||
| 1 |  |
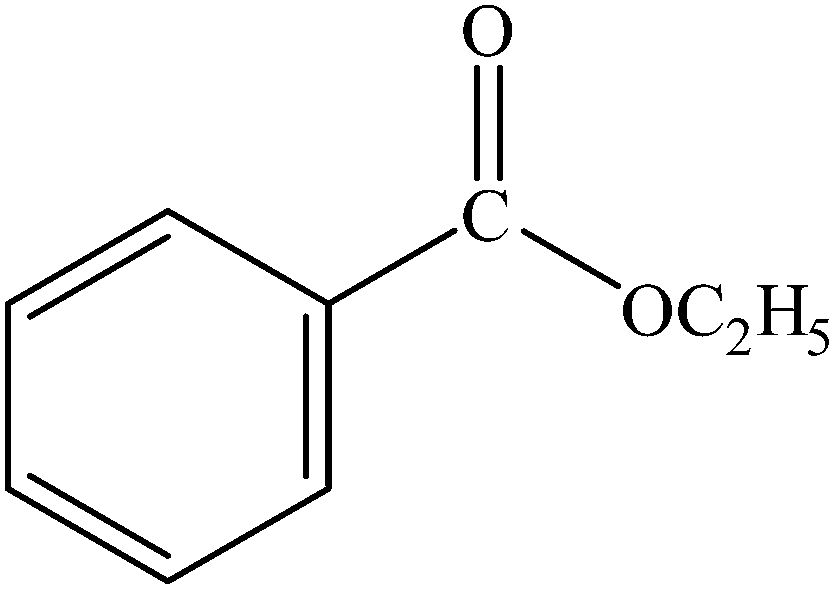 |
96 | 2400 |
| 2 |  |
 |
98 | 2450 |
| 3 |  |
 |
99 | 2475 |
| 4 |  |
 |
98 | 2450 |
| 5 |  |
 |
96 | 2400 |
| 6 |  |
 |
98 | 2450 |
| 7 |  |
 |
98 | 2450 |
| 8 |  |
 |
98 | 2450 |
| 9 |  |
 |
95 | 2375 |
| 10 |  |
 |
96 | 2400 |
| 11 |  |
 |
95 | 2375 |
| 12 |  |
 |
96 | 2400 |
| 13 |  |
 |
96 | 2400 |
| 14 |  |
 |
95 | 2375 |
| 15 |  |
 |
98 | 2450 |
| 16 |  |
 |
70 | 1750 |
| 17 |  |
 |
98 | 2450 |
| 18 |  |
 |
98 | 2450 |
A range of aryl bromides can be converted to the ethyl esters including ortho-substituted examples (Table 4, entries 2, 5 and 8). The yields of different electrophiles varied from 70 to 99%. Functional groups such as methoxy, cyano, ketone, ester, and nitryl groups were well tolerated (Table 4, entries 5–9, 12–14). Fluoro and trifluoromethyl group at para – positions were also effectively converted into their corresponding carbonylative products in excellently high yields (Table 4, entries 10 and 11). As representatives of hetero-aromatic substrates, 3-bromopyridine, gave a high yield of the desired ester (Table 4, entry 15) while 2-bromothiophene only resulted in moderate yield, probably because of the volatility of the product (Table 4, entry 16). 2-Bromonaphthalene and biaryl compound 4-bromobiphenyl was effectively converted (Table 4, entries 17 and 18).
Aminocarbonylation reaction of aryl bromide and amine catalyzed by PS-AMP-Pd
The scope of PS-AMP-Pd was further extended for aminocarbonylation of aryl bromides with a range of aliphatic, aromatic, primary, and secondary amines (Scheme 4). | ||
| Scheme 4 Aminocarbonylation of aryl bromides with various amines. | ||
The reaction of bromobenzene with aniline was selected as a model reaction for optimization. The influence of solvent and reaction temperature on the catalytic performance of this system was investigated by employing various solvents temperature (Table 5). Various solvents, such as DMF (90%), water (85%), anisole (64%), and toluene (96%), were screened for the reaction, but toluene was found to be the best solvent. Similarly, various temperatures were screened to get the best result (Table 5).
| Entry | Solvent | Temperature (°C) | Conversion (%) |
|---|---|---|---|
| a Reaction conditions: bromobenzene (1 mmol), aniline (2 mmol), PS-AMP-Pd (4 × 10 −4 mmol), Et3N (3 mmol), solvent (10 mL). | |||
| 1 | DMF | 90 | 90 |
| 2 | Water | 90 | 85 |
| 3 | Anisole | 90 | 64 |
| 4 | Toluene | 70 | 68 |
| 5 | Toluene | 80 | 75 |
| 6 | Toluene | 90 | 96 |
| 7 | Toluene | 100 | 96 |
Further reaction was optimized with respect to other reaction parameters, including time and CO pressure and the best optimized reaction conditions were then applied for the aminocarbonylation of a variety of bromoaryls and amines (Table 7, entries 1–13). The conversion of amino carbonylation is dependent on the CO pressure applied. The effect of the amount of CO on the amino carbonylation of bromobenzene into corresponding amide was studied and the results are shown in Table 6. The influence of reaction time was also investigated by performing the reaction at a time range from 5 h to 10 h with all other parameters fixed. The results are given in Table 6 which reveals that the conversion is dependent on reaction time and maximum conversion was recorded at 9 h. Therefore, 9 h was selected as the optimum reaction time.
| Entry | CO Pressure (atm) | Reaction time (h) | Conversion (%) |
|---|---|---|---|
| a Reaction conditions: bromobenzene (1 mmol), aniline (2 mmol), PS-AMP-Pd (4 × 10 −4 mmol), Et3N (3 mmol), toluene (10 mL), temp (90 °C). | |||
| 1 | 1 | 9 | 55 |
| 2 | 2 | 9 | 72 |
| 3 | 3 | 9 | 96 |
| 4 | 4 | 9 | 96 |
| 5 | 3 | 5 | 49 |
| 6 | 3 | 7 | 74 |
| 7 | 3 | 10 | 96 |
Various substituted aryl and heteroaryl bromides reacted with aromatic and aliphatic amines to give the desired amides in good to excellent yields. The substitution of electron withdrawing and electron donating groups on the aromatic ring of aryl bromides did not have any appreciable influence on the outcome of the reaction. Aryl bromides bearing either electron-donating or electron-withdrawing substituents, afforded the corresponding carbonylative products in excellent yields. Strong electron donating groups on aryl halides are generally known to slow down the oxidative addition step of the catalytic cycle50 and therefore give lower yields (Table 7, entries 7, 16). The model reaction of bromobenzene with aniline provides a 96% yield of the desired product under optimized reaction conditions (Table 7, entry 1). 4-Methoxyaniline reacts with bromobenzene, providing 93% yield of the desired product (Table 7, entry 2). Furthermore, aliphatic amines, such as benzyl amine and cyclohexyl amine, provide excellent yields of the corresponding product (Table 7, entries 3 and 4). 3-(Trifluoromethyl) aniline provides a moderate yield of the desired product (Table 7, entry 5). A variety of meta- and para-electron deficient and – electron-rich aryl bromides gave excellent yields ranging from 90–99% (Table 7, entries 6–9). 1-Bromonaphthalene furnishes 86% yield of the corresponding amide product (Table 7, entry 10). Heteroaryl bromides 3-bromopyridine and 3-bromothiophene also performed well in this reaction (Table 7, entries 11 and 12). Secondary amines, such as N-methyl aniline and morpholine were also tested with aryl bromides for the synthesis of the corresponding carbonylative products (Table 7, entries 13–17).
| Entry | Aryl bromides | Amines | Product | Yieldb (%) | TON |
|---|---|---|---|---|---|
| a Reaction conditions: aryl bromide (1 mmol), amine (2 mmol), PS-AMP-Pd (4 × 10 −4 mmol), Et3N (3 mmol), toluene (10 mL), CO (3 atm) pressure, temp (90 °C), time (7 h).b Isolated yield. | |||||
| 1 |  |
 |
 |
96 | 2400 |
| 2 |  |
 |
 |
93 | 2325 |
| 3 |  |
 |
 |
96 | 2400 |
| 4 |  |
 |
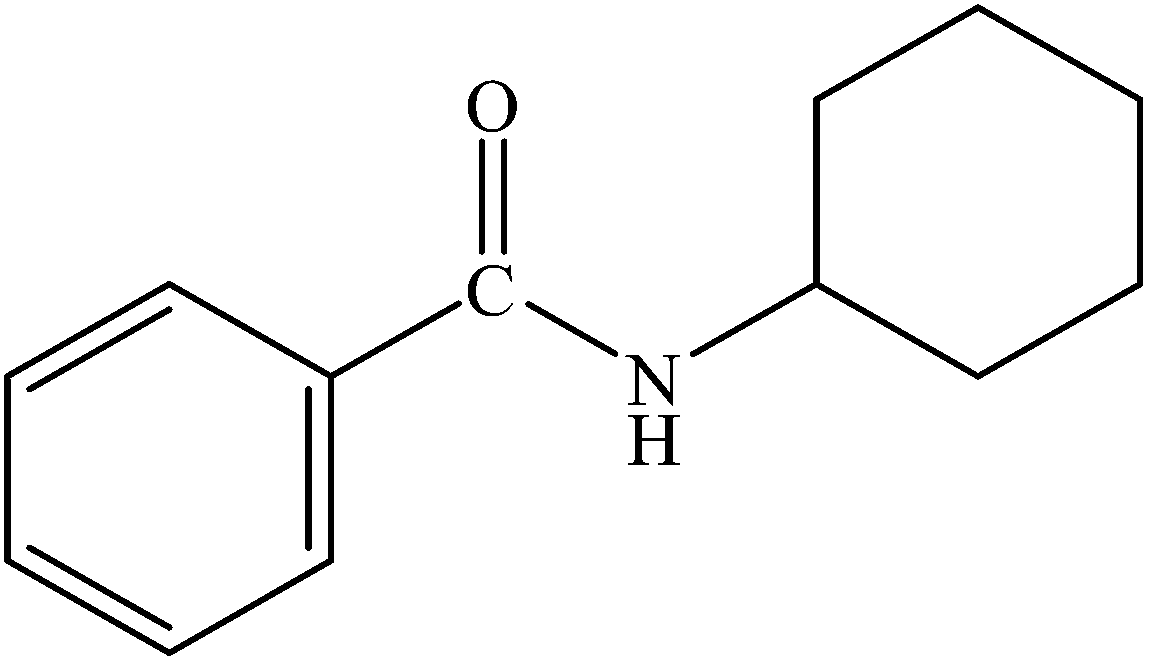 |
98 | 2450 |
| 5 |  |
 |
 |
82 | 2050 |
| 6 |  |
 |
 |
97 | 2425 |
| 7 |  |
 |
 |
90 | 2250 |
| 8 |  |
 |
 |
90 | 2250 |
| 9 |  |
 |
 |
99 | 2475 |
| 10 |  |
 |
 |
86 | 2150 |
| 11 |  |
 |
 |
96 | 2400 |
| 12 |  |
 |
 |
98 | 2450 |
| 13 |  |
 |
 |
97 | 2425 |
| 14 |  |
 |
 |
92 | 2300 |
| 15 |  |
 |
 |
94 | 2350 |
| 16 |  |
 |
 |
88 | 2200 |
| 17 |  |
 |
 |
95 | 2375 |
Test for heterogeneity
The leaching of palladium from polymer anchored palladium(II) complex was confirmed by carrying out analysis of the used catalyst (EDX, IR) as well as the product mixtures (AAS, UV-vis). For AAS analysis, the sample PS-AMP-Pd (0.1 g) was digested with minimum amount of conc. H2SO4 and few drops of hydrogen peroxide. Then it was made up to the mark of 100 mL in volumetric flask with millipore water. In this way stock solution was prepared. Then four standard solution of palladium complex was prepared. From the calibration curve the unknown amount was determined. The same procedure was followed for the sample preparation of the reused catalyst. Analysis of the used catalyst did not show appreciable loss in the palladium content as compared to the fresh catalyst. IR spectrum of the recycled catalyst was quite similar to that of fresh sample indicating the heterogeneous nature of this complex. Analysis of the product mixtures showed that if any palladium was present it was below the detection limit. The UV-vis spectroscopy was also used to determine the stability of this catalyst. The UV-vis spectra of the reaction solution, at the first run, did not show any absorption peaks characteristic of palladium metal, which indicates that the leaching of metal did not take place during the course of the reaction. The metal content of the recycled catalyst was determined with the help of AAS and it was found that palladium content of the recycled catalyst remained almost unaltered. These observations strongly suggest that the present catalyst is truly heterogeneous in nature.Heterogeneity test for the alkoxycarbonylation of bromobenzene with benzyl alcohol was carried out at 70 °C using PS-AMP-Pd catalyst, then after 1.5 h, the PS-AMP-Pd complex catalyst was filtered off and was allowed to react further. We found that no further reaction occurred after this hot filtration procedure; hence, this experimental finding suggests there is no palladium leaching from the PS-AMP-Pd complex during the progress of a reaction. In addition, to reconfirm this observation, ICP-AES analysis of the reaction mixture was carried out after 1.5 and 3 h, which revealed a below detectable level (below 0.01 ppm) of palladium in solution.
Recycling of catalyst
The catalyst remains insoluble in the present reaction conditions and hence can be easily separated. After completion of the reaction the catalyst was recovered by centrifuge and then washed thoroughly with dichloromethane to remove the substrates from its surface. The recovered catalyst was collected and dried at 100 °C. Alkoxycarbonylation and aminocarbonylation reactions were carried out with the recycled catalyst under the optimized reaction conditions. The catalyst was recycled in order to test its activity as well as stability. The obtained results are presented in Fig. 6. As seen from Fig. 6, the catalyst did not show any appreciable change in its activity which indicates that the catalyst is stable and can be regenerated for repeated use. | ||
| Fig. 6 Recycling efficiency of the polymer anchored Pd(II) complex for alkoxy and amino carbonylation reactions. | ||
Conclusions
A polymer anchored palladium Schiff base complex was prepared and characterized. The complex was an active and reusable catalyst for the alkoxycarbonylation and aminocarbonylation reactions. The catalyst is stable and recyclable under the used reaction conditions. This heterogeneous catalyst shows no significant loss of activity in recycling experiments. The active sites do not leach out from the support and thus can be reused without appreciable loss of activity, indicating effective anchoring. The reusability of this catalyst is high without significant decrease in its initial activity.Acknowledgements
SMI acknowledges Department of Science and Technology (DST) and Council of Scientific and Industrial Research (CSIR), New Delhi, India for funding. RAM acknowledges UGC, New Delhi, India for his Maulana Azad National Fellowship.References
- A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed.
- (a) X. F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986–5009 RSC; (b) M. Seayad, J. Seayad, P. L. Mills and R. V. Chaudhari, Ind. Eng. Chem. Res., 2003, 42, 2496–2506 CrossRef; (c) S. P. Gupte, V. P. Krishnamurthy and R. V. Chaudhari, Chem. Eng. Sci., 1996, 51, 2069–2078 CrossRef CAS.
- H. Neumann, A. Brennführer and M. Beller, Chem.–Eur. J., 2008, 14, 3645–3652 CrossRef CAS PubMed.
- Z. Zhang, Y. Liu, M. Gong, X. Zhao, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 1139–1142 CrossRef CAS PubMed.
- X. F. Wu, H. Neumann, A. Spannenberg, T. Schulz, H. Jiao and M. J. Beller, J. Am. Chem. Soc., 2010, 132, 14596–14602 CrossRef CAS PubMed.
- Q. Liu, G. Li, J. He, J. Liu, P. Li and A. Lei, Angew. Chem., Int. Ed., 2010, 49, 3371–3374 CrossRef CAS PubMed.
- X. F. Wu, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 5284–5287 CrossRef CAS PubMed.
- T. Sugihara, C. Coperet, Z. Owczarcy, L. S. Haring and E. J. Negishi, J. Am. Chem. Soc., 1994, 116, 7923–7924 CrossRef CAS.
- M. V. Khedkar, T. Sasaki and B. M. Bhanage, ACS Catal., 2013, 3, 287–293 CrossRef CAS.
- K. Ishihara, Tetrahedron, 2009, 65, 1085–1109 CrossRef CAS PubMed.
- M. Beller, B. Cornils, C. D. Frohning and C. W. J. Kohlpaintner, J. Mol. Catal. A: Chem., 1995, 104, 17–85 CrossRef CAS.
- J. Wannberg and M. Larhed, J. Org. Chem., 2003, 68, 5750–5753 CrossRef CAS PubMed.
- H. U. Blaser, M. Diggelmann, H. Meier, F. Naud, E. Scheppach, A. Schnyder and M. Studer, J. Org. Chem., 2003, 68, 3725–3728 CrossRef CAS PubMed.
- C. M. Kormos and N. E. Leadbeater, Org. Biomol. Chem., 2007, 5, 65–68 CAS.
- J. Liu, B. Liang, D. Shu, Y. Hub, Z. Yang and A. Lei, Tetrahedron, 2008, 64, 9581–9584 CrossRef CAS PubMed.
- E. Mizushima, T. Hayashi and M. Tanka, Green Chem., 2001, 3, 76–79 RSC.
- M. A. Mercadante and N. E. Leadbeater, Org. Biomol. Chem., 2011, 9, 6575–6578 CAS.
- C. B. Kelly, C. Lee, M. A. Mercadante and N. E. Leadbeater, Org. Process Res. Dev., 2011, 15, 717–720 CrossRef CAS.
- J. W. Bode, Curr. Opin. Drug Discovery Dev., 2006, 9, 765–775 CAS.
- T. Cupido, J. Tulla-Puche, J. Spengler and F. Albericio, Curr. Opin. Drug Discovery Dev., 2007, 10, 768–783 CAS.
- B. Katzung, S. Masters and A. Trevor, Basic and Clinical Pharmacology, Mcgraw-Hill, New York, 12th edn, 2011 Search PubMed.
- K. Kumar, A. Zapf, D. Michalik, A. Tillack, T. Heinrich, H. Bottcher, M. Arlt and M. Beller, Org. Lett., 2004, 6, 7–10 CrossRef CAS PubMed.
- P. D. Bailey, T. J. Mills, R. Pettecrew, R. A. Price, A. R. Katritzky and R. J. K. Taylor, in Comprehensive Organic Functional Group Transformations II, Elsevier, Oxford, UK., 2005, vol. 5, pp. 201–292 Search PubMed.
- B. S. Jursic and Z. Zdravkovski, Synth. Commun., 1993, 23, 2761–2770 CrossRef CAS.
- A. L. J. Beckwith and J. Zabicky, Synthesis of Amides, in The Chemistry of Amides, Interscience, London, 1970, pp. 105–109 Search PubMed.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS PubMed.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- P. Xie, C. Xia and H. Huang, Org. Lett., 2013, 15(13), 3370–3373 CrossRef CAS PubMed.
- A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3318–3326 CrossRef CAS.
- W. Magerlein, A. F. Indolese and M. J. Beller, Organomet. Chem., 2002, 641, 30–40 CrossRef CAS.
- A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327–3330 CrossRef CAS.
- P. W. Miller, N. J. Long, A. J. Mello, R. Vilar, J. Passchier and A. Gee, Chem. Commun., 2006, 5, 546–548 RSC.
- J. R. Martinelli, T. P. Clark, D. A. Watson, R. H. Munday and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 8460–8463 CrossRef CAS PubMed.
- J. R. Martinelli, D. A. Watson, D. M. M. Freckmann, T. E. Barder and S. L. Buchwald, J. Org. Chem., 2008, 73, 7102–7107 CrossRef CAS PubMed.
- E. Mizushima, T. Hayashi and M. Tanaka, Green Chem., 2001, 3, 76–79 RSC.
- (a) M. Genelot, N. Villandier, A. Bendjeriou, P. Jaithong, L. Djakovitch and V. Dufaudz, Catal. Sci. Technol., 2012, 2, 1886–1893 RSC; (b) J. Liu, S. Zheng, W. Sun and C. Xia, Chin. J. Chem., 2009, 27, 623–627 CrossRef CAS PubMed; (c) H. Cao and H. Alper, Org. Lett., 2010, 12, 4126–4129 CrossRef CAS PubMed; (d) E. Mizushima, T. Hayashi and M. Tanaka, Green Chem., 2001, 3, 76–79 RSC.
- (a) S. Roy, S. Roy and G. W. Gribble, Tetrahedron, 2012, 68, 9867–9923 CrossRef CAS PubMed; (b) A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327–3331 CrossRef CAS; (c) Y. Uozumi, T. Arii and T. Watanabe, J. Org. Chem., 2001, 66, 5272–5274 CrossRef CAS; (d) S. Ho, G. Bondarenko, D. Rosa, B. Dragisic and A. Orellana, J. Org. Chem., 2012, 77, 2008–2012 CrossRef CAS PubMed; (e) V. Fuente, C. Godard, C. Claver and S. Castillon, Adv. Synth. Catal., 2012, 354, 1971–1979 CrossRef PubMed; (f) Z. S. Qureshi, S. A. Revankar, M. V. Khedkar and B. M. Bhanage, Catal. Today, 2012, 198, 148–153 CrossRef CAS PubMed.
- A. I. Vogel, Text book of practical organic chemistry, Quantitative Analysis, Longman, London, 5th edn, 1998 Search PubMed.
- J. M. Frechet and C. Schuerch, J. Am. Chem. Soc., 1971, 93, 492–496 CrossRef.
- M. Islam, P. Mondal, S. Mukherjee, M. Mobarak, A. S. Roy, S. Mondal and S. Sarkar, J. Chem. Technol. Biotechnol., 2010, 85, 460–470 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, 4th edn, 1986 Search PubMed.
- M. Islam, P. Mondal, K. Tuhina, A. S. Roy, S. Mondal and D. Hossain, J. Organomet. Chem., 2010, 695, 2284–2295 CrossRef CAS PubMed.
- B. V. Patel, K. Desai and T. Thaker, Synth. React. Inorg. Met.–Org. Chem., 1989, 19, 391–395 CrossRef CAS.
- G. G. Mohamed, M. M. Omar and A. H. H. Hindy, Spectrochim. Acta, Part A, 2005, 62, 1140–1150 CrossRef PubMed.
- J. R. Durig, R. Layton, D. W. Sink and B. R. Mitchell, Spectrochim. Acta, 1965, 21, 1367–1378 CrossRef CAS.
- R. Mani, V. Mahadevan and M. Srinivasan, Br. Polym. J., 1990, 22, 177–184 CAS.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier Science Publication, New York, 2nd edn, 1986 Search PubMed.
- E. Bermejo, R. Carballo, A. Castineirs, R. Dominguez, A. E. Liberta, C. M. Mossmer, M. M. Salberg and D. X. West, Eur. J. Inorg. Chem., 1999, 965–973 CrossRef CAS.
- (a) A. M. Sajith and A. Muralidharan, Tetrahedron Lett., 2012, 53, 1036–1041 CrossRef CAS PubMed; (b) A. M. Sajith and A. Muralidharan, Tetrahedron Lett., 2012, 53, 5206–5210 CrossRef CAS PubMed; (c) J. F. Hartwig, Inorg. Chem., 2007, 46, 1936–1947 CrossRef CAS PubMed; (d) V. F. Slagt, A. H. M. De Vries, J. G. De Vries and R. M. Kellogg, Org. Process Res. Dev., 2010, 14, 30–47 CrossRef CAS; (e) P. Mondal, S. Banerjee, A. S. Roy, T. K. Mandal and S. M. Islam, J. Mater. Chem., 2012, 22, 20434–20442 RSC; (f) S. M. Islam, P. Mondal, A. S. Roy, S. Mondal and D. Hossain, Tetrahedron Lett., 2010, 51, 2067–2070 CrossRef CAS PubMed.
- (a) S. T. Gadge and B. M. Bhanage, Org. Biomol. Chem., 2014, 12, 5727–5732 RSC; (b) T. T. Dang, Y. Zhu, S. C. Ghosh, A. Chen, C. L. L. Chai and A. M. Seayad, Chem. Commun., 2012, 48, 1805–1807 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra05365f |
| ‡ These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2014 |