
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermoresponsive biopolymer hydrogels with tunable gel characteristics
Erfan
Dashtimoghadam
ab,
Hamid
Mirzadeh
*a,
Faramarz Afshar
Taromi
a and
Bo
Nyström
*b
aDepartment of Polymer Engineering and Color Technology, Amirkabir University of Technology, 424 Hafez Avenue, Tehran, Iran. E-mail: mirzadeh@aut.ac.ir
bDepartment of Chemistry, University of Oslo, P.O. Box 1033, Blindern, N-0315 Oslo, Norway. E-mail: bo.nystrom@kjemi.uio.no; Tel: +47-22855522
First published on 27th August 2014
Abstract
Here we describe the design of thermosensitive biopolymer-based hydrogels with adjustable gel properties. It is demonstrated that formulations comprising predetermined contents of hydrophobically modified chitosan biopolymer with n-dodecyl groups (HC) undergo a quick fine-tunable gelation in the presence of β-glycerophosphate disodium salt (GP) triggered by increasing temperature. In-depth rheological characterizations revealed that with increasing HC content in solutions, the gel point shifts to lower temperatures as more compact networks with considerably improved gel strength are formed. The evolution of hydrogels was also investigated by the small angle light scattering (SALS) technique. It was realized that the incorporation of HC chains induce a gel to be accomplished through formation of smaller but more homogenous microdomains with the aid of pendant hydrophobic moieties in the lower temperature range. Moreover, time sweep rheological characterization of the thermogelling systems disclosed that the gelation process proceeds faster in the presence of HC chains. The Fredrickson–Larson (F–L) theory was implemented as an alternative approach to determine the gel point. In view of the obtained results which were consistent with the classical Winter–Chambon theory, the F–L theory can be proposed as a robust and less tedious method to precisely determine the gel point in such systems. The developed hydrogels can be proposed as promising injectable matrices with predetermined gel features for various biomedical applications.
1. Introduction
Hydrogels represent an appealing class of materials in the biomaterials research community.1 In view of biophysical similarities to soft tissues, inherent biological adaptability, potential responsiveness to environmental changes and ability to encapsulate specific functionalities, hydrogels have emerged as perfect biomimetic three-dimensional matrices for widespread life science applications, including regenerative medicine and drug delivery systems.2,3Stimuli responsive hydrogels are becoming increasingly important in the development of intelligent carriers.4,5 Injectable hydrogels that can be formed in response to a certain stimulus (e.g., temperature) offer interesting opportunities like delivery of bioactive and therapeutic agents in a minimally invasive manner.6In situ thermogelling hydrogels undergo sol–gel phase transition above the lower critical solution temperature (LCST), which is typically predetermined to be around the body temperature. These systems present promising features like the lack of organic solvents, cross-linkers, and elimination of adverse thermal alterations on surrounding tissues.7–10
Among different proposed macromolecules in the processing of hydrogel matrices, naturally derived polymers have become the focus of attention as promising candidates.11,12 Biodegradability, biocompatibility, chemical versatility, similarity with the extracellular matrix, and intrinsic cellular interaction are considered as their peculiar bio-characteristics for biomedical applications. Naturally derived polymers like polysaccharides may also avoid the stimulation of chronic inflammation or immunological reactions and toxicity.13–15 Chitosan, as the deacetylated and cationic derivative of chitin, has invoked an increasing interest owing to its desirable properties including structural similarity to naturally occurring glycosaminolglycans, biofunctionality and non-antigenicity.16–18 In addition, amino and hydroxyl groups on chitosan backbone provide chemical modification versatility.19,20 It has been shown that neutralization of acidic chitosan formulations with glycerol phosphate disodium salt (GP) results in its thermogelation at physiological pH range.21,22 The most important interactions responsible for this thermally induced sol–gel transition are the hydrophobic interactions among chitosan chains, enhanced by the structuring action of glycerol on water molecules.23
Before hydrogelation, cells or bioactive agents can be incorporated into the polymer solutions to provide practical tissue engineering or pharmaceutical applications. As the features of hydrogels play substantial role in biomedical applications such as extracellular microenvironments,24,25 our objective is to investigate the effect of substituted hydrophobic moieties (i.e., n-dodecyl groups) onto chitosan chains on gel properties of their mixtures with GP. It should be noted that in spite of developed various chitosan/GP thermogelling systems, design of hydrogels with adjustable gel characteristics through modulation of hydrophobic interactions at constant content of salt has not been reported so far. In this work, we intend in a controlled way to tune the gel characteristics including sol–gel transition behavior and gel strength through predetermined incorporation of hydrophobically modified chitosan chains into the developed thermoresponsive blend formulations.
2. Experimental section
2.1. Materials and preparation of solutions
Chitosan (deacetylation degree of 84% and a weight-average molecular weight of ∼4 × l05) was acquired from Pronova Biopolymers. Glycerol-2-phosphate disodium salt hydrate was supplied by Merck. The chemical modification of chitosan was carried out through the reaction of amino groups with a C12-aldehyde. The substitution of C12 aliphatic hydrophobic groups on chitosan was confirmed by 1H NMR spectroscopy (Bruker 400 MHz). The details of the characterization methods and modification procedure have been described before.26,27 The substitution degree of the hydrophobically modified chitosan was determined by ninhydrin method.28,29 Briefly described, polymer was dissolved in aqueous acetic acid solution, followed by adding 0.5 mL of 4 M acetic acid–acetate buffer (pH = 5.5) into 0.5 mL of solution. Then, 1 mL of ninhydrin reagent (Sigma-Aldrich) was added and test tube was incubated in a boiling water bath for 20 min. The solution was subsequently cooled to room temperature and the absorbance was measured at 570 nm (Shimadzu UVmini-1240 UV-Vis spectrophotometer). The unmodified chitosan was used as the control sample and the acetic acid–acetate buffer solution was used as the blank. Accordingly, hydrophobic substitution degree for the sample denoted as HC was determined to be 2.3%.To prepare unmodified chitosan/β-glycerophosphate disodium salt (UC/GP) solution as well as the blend formulations comprising hydrophobically modified analogue (UC-HC/GP), UC was first dissolved and mixed with various amounts of HC solutions. The prepared solutions were then chilled to 4 °C, and subsequently cold GP solution in water was added drop-wise under magnetic stirring to adjust the pH of the polymeric solutions to the physiological range. All formulations were comprising constant total content of polymer (2 wt%) and GP (0.78 M) concentration, but different HC loading (0, 0.5, and 1 wt%). Accordingly, the samples were assigned as UC2−x–HCx/GP, where x is the weight percentage of the HC in the blend formulations. It should be mentioned that to retain the injectability of the solutions, the content of the HC was not exceeded 1 wt%.
2.2. Rheological measurements
Oscillatory shear experiments were performed in an Anton Paar-Physica MCR 301 rheometer using a cone-and-plate geometry, with a diameter of 75 mm and a cone angle of 1°. The measuring apparatus is equipped with a temperature unit (Peltier plate), which provides an effective temperature control (±0.05 °C) for an extended time over the studied temperature range. The free surface of solutions was covered with a thin layer of low-viscosity silicone oil to prevent the dehydration of the samples. However, the viscosity value is practically not affected by the oil layer. All measurements were performed at a heating rate of 1 K min−1. Strain amplitude of 3% was applied to minimize the perturbation of the network during the gel evolution process. Before the rheological measurements, the strain amplitude was checked to ensure the measurements are conducted within the linear viscoelastic regime and the dynamic storage modulus (G′) and loss modulus (G′′) to be independent of the strain amplitude.2.3. Turbidimetry
Turbidity experiments were carried out by means of an NK60-CPA cloud point analyzer from Phase Technology, Richmond, BC, Canada. The details of this equipment have been described elsewhere.30 This operation of the device is based on a scanning diffusive technique to characterize phase changes of the sample with high accuracy and sensitivity. The light beam from an AlGaAs light source, operating at 654 nm, is focused on the measuring sample applied onto a glass plate coated with a thin metallic layer of very high reflectivity. Directly above the sample, an optical system with a light-scattering detector continuously monitors the scattered intensity signal (S) of the sample as it is subjected to prescribed temperature alterations. The relation between the signal and the turbidity (τ) is given by the empirical equation, τ(cm−1) = 9 × 10−9S3.751.30 The heating rate was set to 1 °C min−1 in the turbidity measurements.2.4. Small angle light scattering (SALS)
SALS measurements were performed using a CCD camera (driver LuCam V. 3.8), placed in a parallel position to the screen, and captured two-dimensional images from the scattering patterns of the samples. Time exposure for the acquisition of images was 200 ms. Samples were incubated between a specially designed plate–plate glass system. Plate diameter was 43 mm and the gap between the plates was 0.25 mm. Temperature was gradually changed from 10 to 45 °C. A detailed description of this technique has been provided in an earlier publication.313. Results and discussion
3.1. Analysis of rheological data and investigation of gel properties
To gain insights into the influence of incorporated hydrophobically modified chitosan chains on the resultant gel characteristics, rheological behavior of the systems in the course of gelation beyond the gel point was scrutinized. The gelation temperature of the prepared formulations was determined by frequency-independent value of the loss tangent (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ = G′′/G′) obtained from a multi-frequency plot versus temperature (Fig. 1).32 A common feature observed in Fig. 1 for all thermogelling solutions is that the damping factor is frequency dependent and decreases during the gel formation, indicating gradually enhanced elasticity of the studied systems with increasing temperature.
δ = G′′/G′) obtained from a multi-frequency plot versus temperature (Fig. 1).32 A common feature observed in Fig. 1 for all thermogelling solutions is that the damping factor is frequency dependent and decreases during the gel formation, indicating gradually enhanced elasticity of the studied systems with increasing temperature.
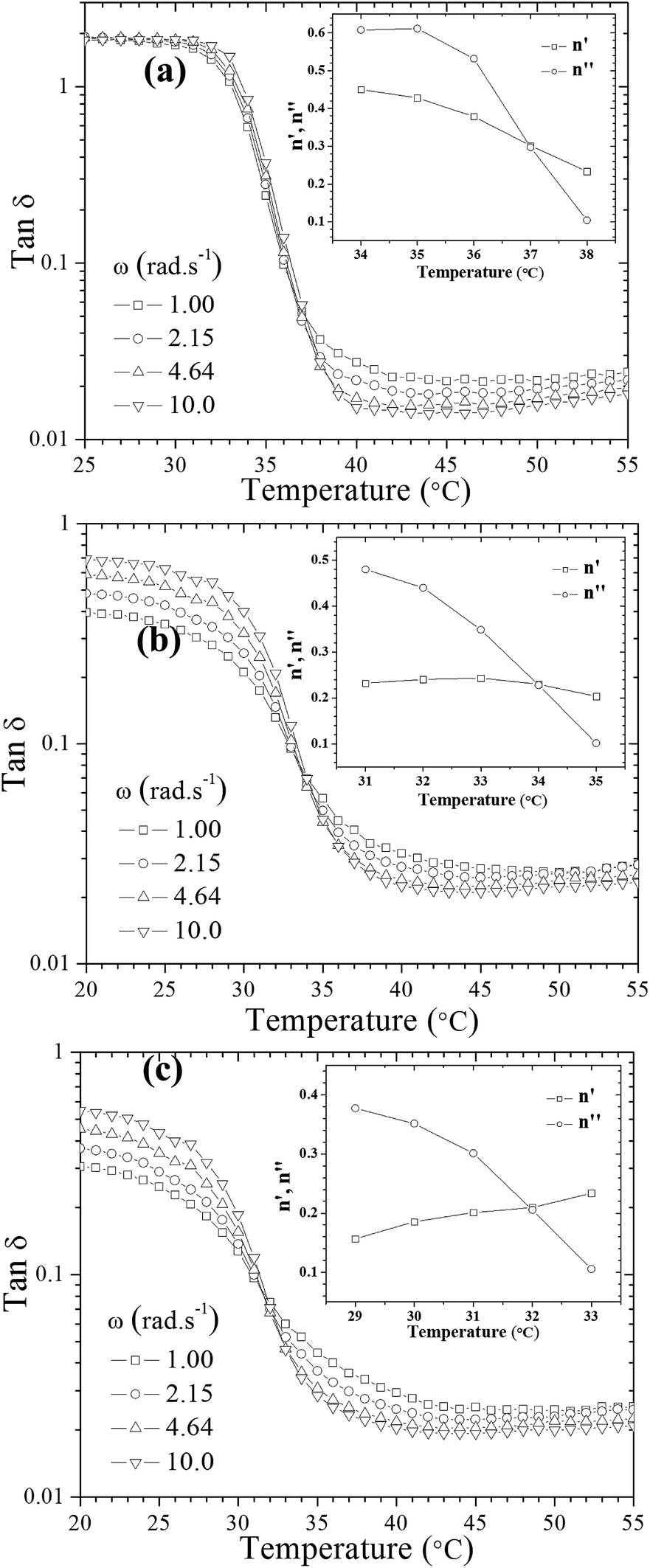 | ||
| Fig. 1 Illustration of methods for the determination of the gel point: viscoelastic loss tangent as a function of temperature at the indicated frequencies for (a) UC2.0–HC0.0/GP, (b) UC1.5–HC0.5/GP, (c) UC1.0–HC1.0/GP. The inset plot shows changes of apparent exponents (n′ for storage and n′′ for loss moduli) at various temperatures. | ||
An alternative method was also employed to determine the gel point33,34 which is based on crossover of the apparent viscoelastic exponents n′ and n′′(G′ ∼ ωn′; G′′ ∼ ωn′′) calculated from the frequency dependence of G′ and G′′ at different temperatures (see the inset graphs in Fig. 1a–c). Both methods resulted in the same gelation temperatures of 37, 34 and 32 °C for UC2.0–HC0.0/GP, UC1.5–HC0.5/GP, and UC1.0–HC1.0/GP solutions, respectively. As seen, the higher content of HC chains in the formulations, the lower incipient gelation temperature, implying the effective contribution of HC chain in the developing gel networks. As a matter of fact, addition of GP into chitosan solutions modulates molecular forces including hydrogen bonds, electrostatic and hydrophobic interactions involved in the consequent thermogelation process. In view of this and considering water-structuring character of GP molecules22,23 hydrophobic interactions are enhanced with increasing temperature; hence, in the presence of HC chains, sol–gel transition has been facilitated and expedited through strengthened hydrophobic attractions in a controllable manner. The molecular interactions involved in gelation of the chitosan–hydrophobically modified chitosan/β-glycerophosphate blend thermoresponsive formulations are displayed in Scheme 1.
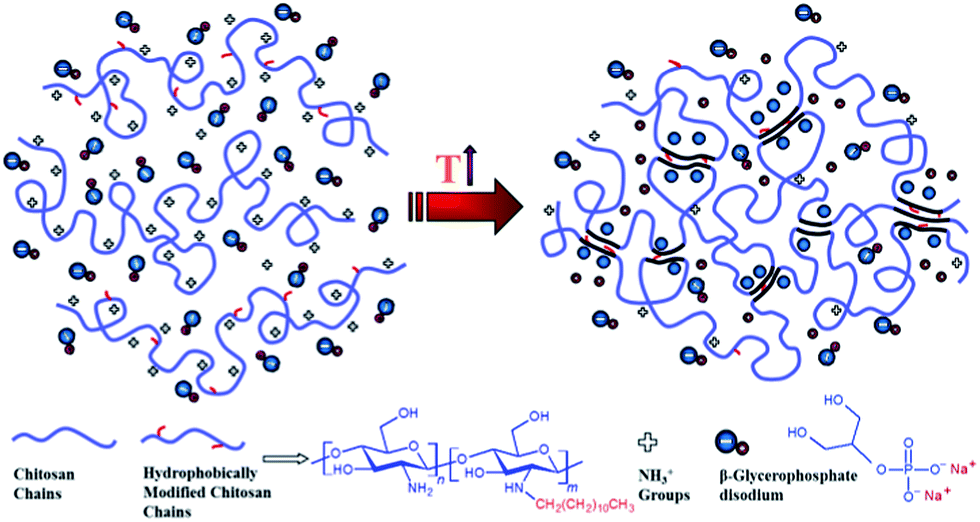 | ||
| Scheme 1 Illustration of molecular interactions involved in gelation of chitosan (UC)–hydrophobically modified chitosan (HC)/β-glycerophosphate (GP) thermoresponsive formulations. | ||
It is worth to notice that, though the gelation temperature of the chitosan/GP solutions can be altered with GP content, but here we presented a new approach to govern gelation of such systems without increasing GP to maintain their biocompatibility.
As described above, the values of relaxation exponent are determined from a log–log plot and scaling relation of storage and loss modulus at the gel point (G′(ω) ∝ G′′(ω) ∝ ωn) (cf.Fig. 2a–c). The evolution of the loss and storage modulus for UC2.0–HC0.0/GP and UC1.0–HC1.0/GP solutions during the heating cycle has been displayed in Fig. 3. As seen, for both systems the gelation proceeds through three main regimes: (i) an inductive behavior at low temperatures, (ii) a rapid gelation in the vicinity of the gel point, wherein a steep increase in the moduli is observed, (iii) and a slow gelation process at higher temperatures. At lower temperatures, the UC1.0–HC1.0/GP system shows higher elasticity highlighting the increased hydrophobic intermolecular interactions. It is also worth noting that tanδ of the blend systems is less than 1 at all stages of gelation, which again indicates the effective contribution of incorporated conjugated hydrophobic groups on interactions among chitosan chains (Fig. 1b and c).
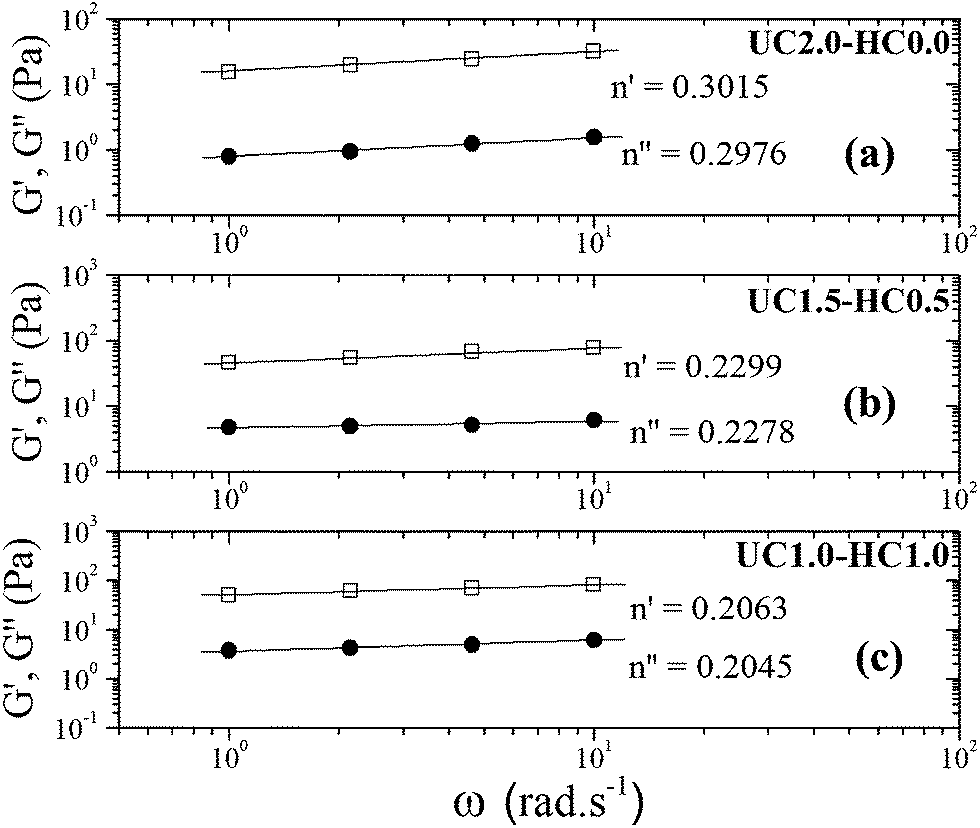 | ||
| Fig. 2 The power law behavior of the dynamic moduli for 2 wt% incipient gels of (a) UC2.0–HC0.0/GP at 37 °C, (b) UC1.5–HC0.5/GP at 34 °C, and (c) UC1.0–HC1.0/GP at 32 °C. | ||
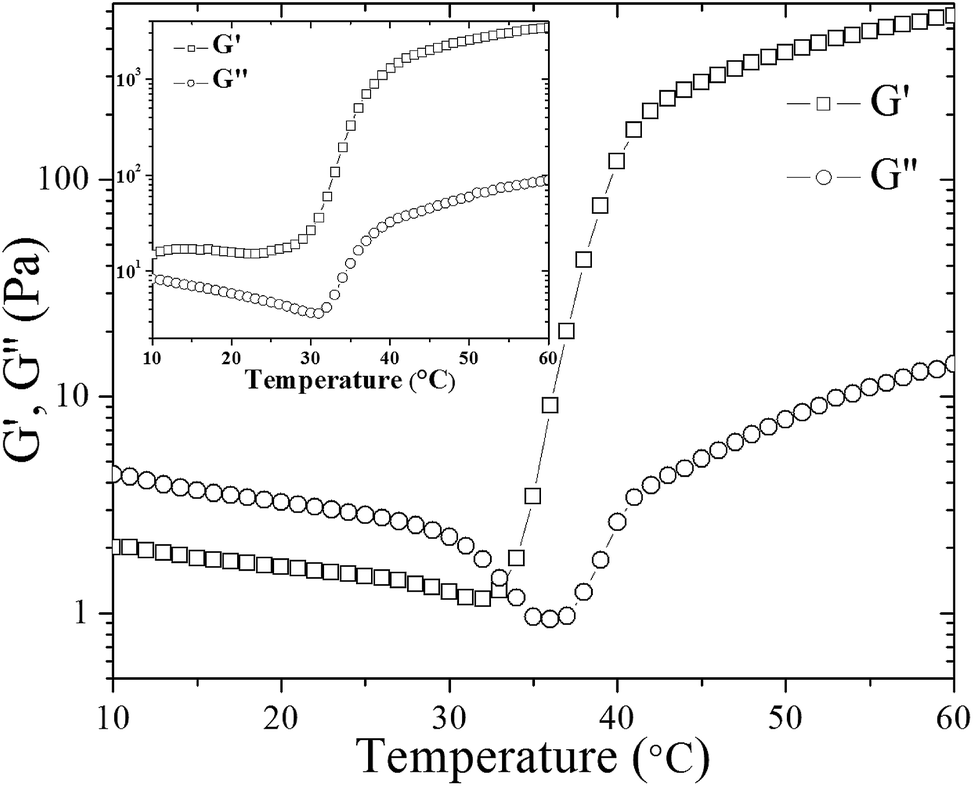 | ||
| Fig. 3 The evolution of storage (G′) and loss (G′′) modulus of UC2.0–HC0.0/GP sample during heating from 10 to 60 °C. Inset shows the corresponding graph for UC1.0–HC1.0/GP sample. | ||
Nonetheless, considering the shear thinning behavior of such systems,27 diffusion of the hydrogel precursors into the surrounding tissues is favorably eliminated for biomedical applications.
It has been shown that water molecules around chitosan chains are removed upon heating, and a heat induced transfer of protons to glycerophosphate groups from protonated amino groups occurs; thus, hydrophobic contacts among chitosan chains are enhanced.35 Actually, the formation of such hydrophobic interactions is accompanied by G′ and G′′ crossover in the case of UC2.0–HC0.0/GP, which here occurred at 33 °C (Fig. 3).
Although in some studies the G′ and G′′ crossover has been considered to determine the gel point,36 but this definition is not commonly a reliable criterion for determination of the gel point.32 In fact, theoretical calculations have shown that the G′ = G′′ criterion define the gelation conditions only for a particular relaxation behavior (n = 1/2).37 Accordingly, G′ and G′′ crossover point for UC2.0–HC0.0/GP in Fig. 3, which is not observed for UC1.0–HC1.0/GP, can be attributed to the formation of hydrophobic domains as the first stage of gelation.
Gel strength of the designed formulations was calculated based on the Winter–Chambon theory.32 The gel strength parameter was determined according to eqn (1) for the incipient gels:
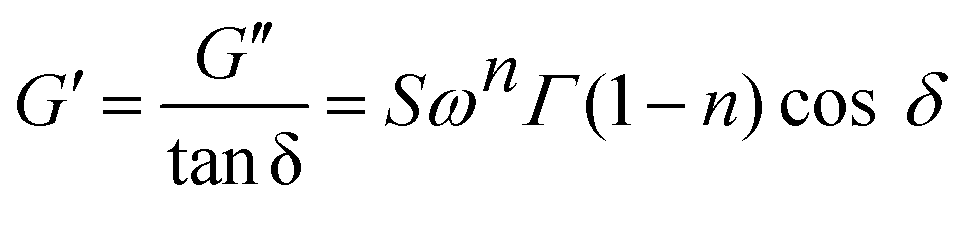 | (1) |
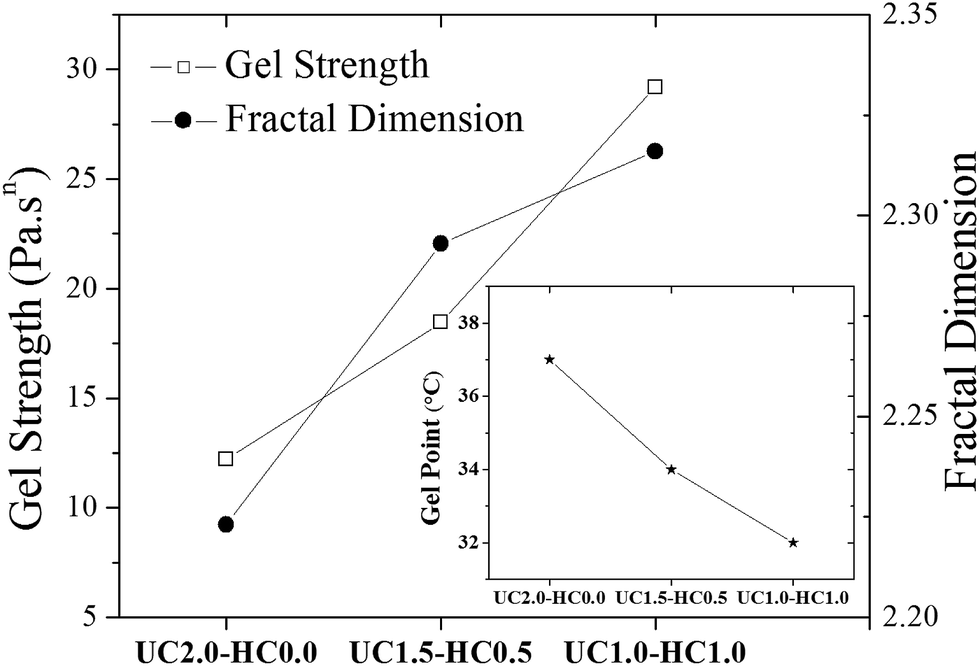 | ||
| Fig. 4 The gel strength parameter (see eqn (1)) and the fractal dimension (see eqn (2)) are shown for the indicated incipient gelling systems. The inset plot shows changes of the gel temperature as the content of hydrophobically modified chitosan chains in the formulation is changed. | ||
Muthukumar elaborated a theoretical approach,38 based on the assumption that strand length variations between crosslinks in the incipient gel network give rise to changes of the excluded volume interactions, to rationalize values of the relaxation exponent in the whole physically accessible range (0 < n < 1). In this model, a relationship between the fractal morphology of the incipient gel network and n was expressed through eqn (2):
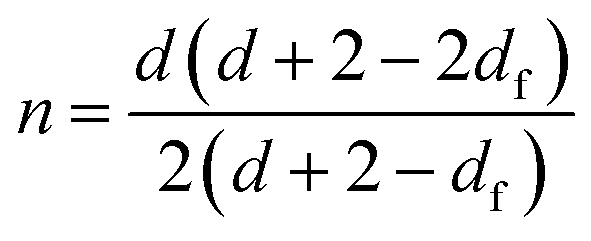 | (2) |
As mentioned above, an incipient gel is not a fully developed network and at this point, macromolecules involved in gel network coexist with and are swelled by the free molecules of finite structures (sol). Accordingly, formation of spatially bicontinuous percolated networks is considered as the decisive point for gelation. Since the gelation process can be phenomenologically considered as analogous to a phase separation process, some of the present authors have recently implemented the Frederickson–Larson (F–L) theory to study sol–gel transitions of thermosensitive chitosan/GP systems; the percolation structuring temperature was determined.39 Near the phase separation temperature of polymer blends or the order–disorder transition in block copolymers, anomalous viscoelastic behavior is observed. Fredrickrson and Larson developed a mean-field theory to derive the contribution to the shear stress caused by critical fluctuations near the order–disorder transition.40 They have demonstrated that extra stress induced by critical fluctuations can be calculated from the total free energy differential change and after integration over the whole wave vector space, eqn (3) and (4) for the dynamic storage and loss modulus, respectively, can be expressed as:
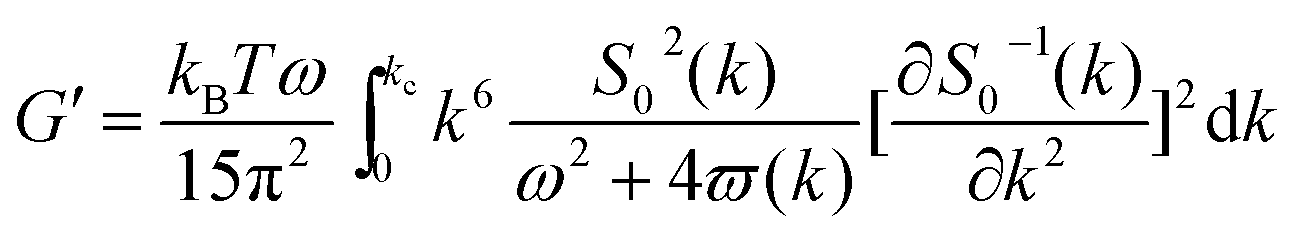 | (3) |
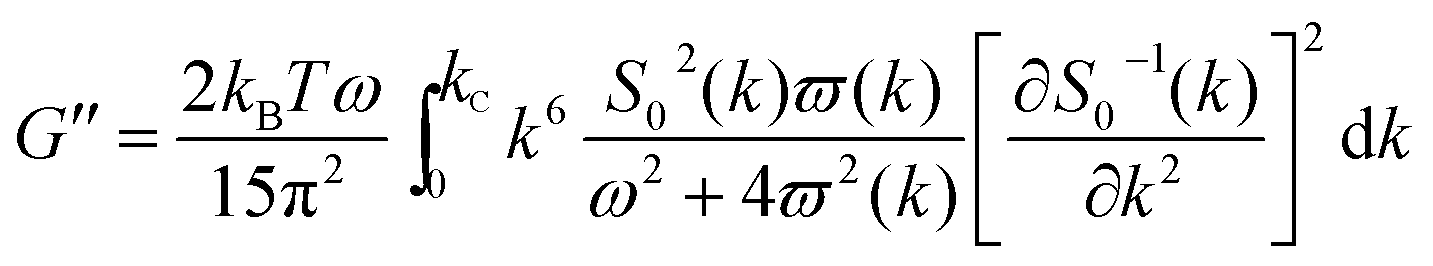 | (4) |
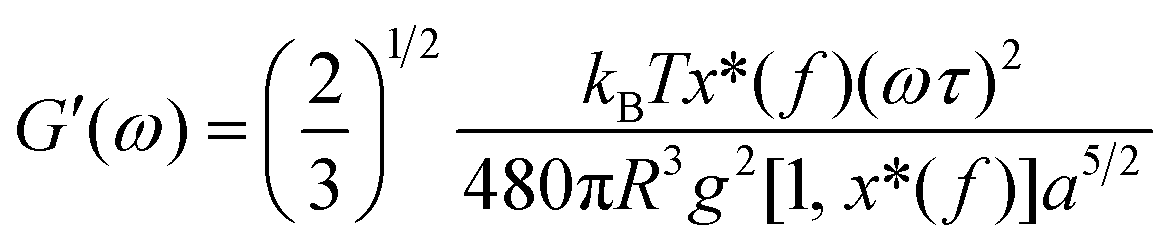 | (5) |
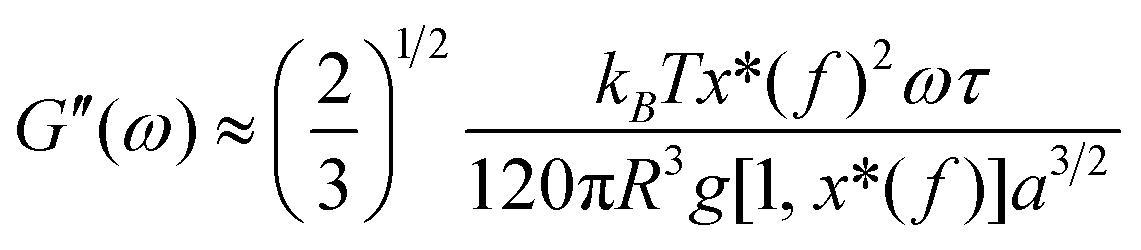 | (6) |
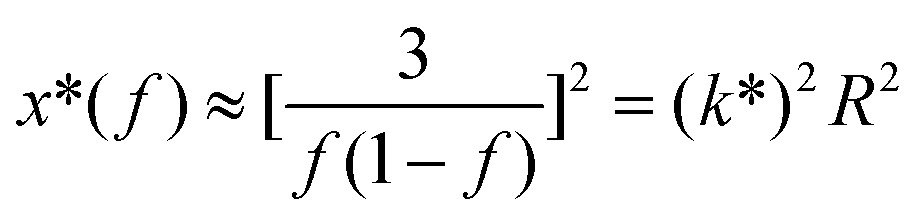 where S has its maximum. Thus, the
where S has its maximum. Thus, the 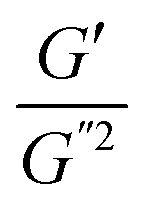 ratio is calculated with the aid of eqn (7):
ratio is calculated with the aid of eqn (7):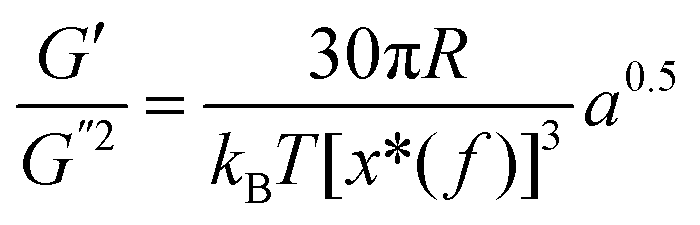 | (7) |
Based on eqn (7), (G′T/G′′2)2 shows a divergence near the transition temperature. Fig. 5 displays the determination of the gel point based on F–L theory. The results show that the gel temperatures determined in the framework of this approach for the UC/GP and UC–HC/GP blend systems are in good agreement with those obtained from the Winter–Chambon theory. The consistency in the determination of the gel-points for these complex systems by using two approaches of different origin reveals that the procedures are robust and they work also for systems with more intricate mechanisms of sol–gel transitions. In view of this, the F–L theory can be proposed as a less tedious method (which has been implemented here based on temperature sweep viscoelastic properties at just one angular frequency) to precisely determine the gel point of the studied thermogelling systems.
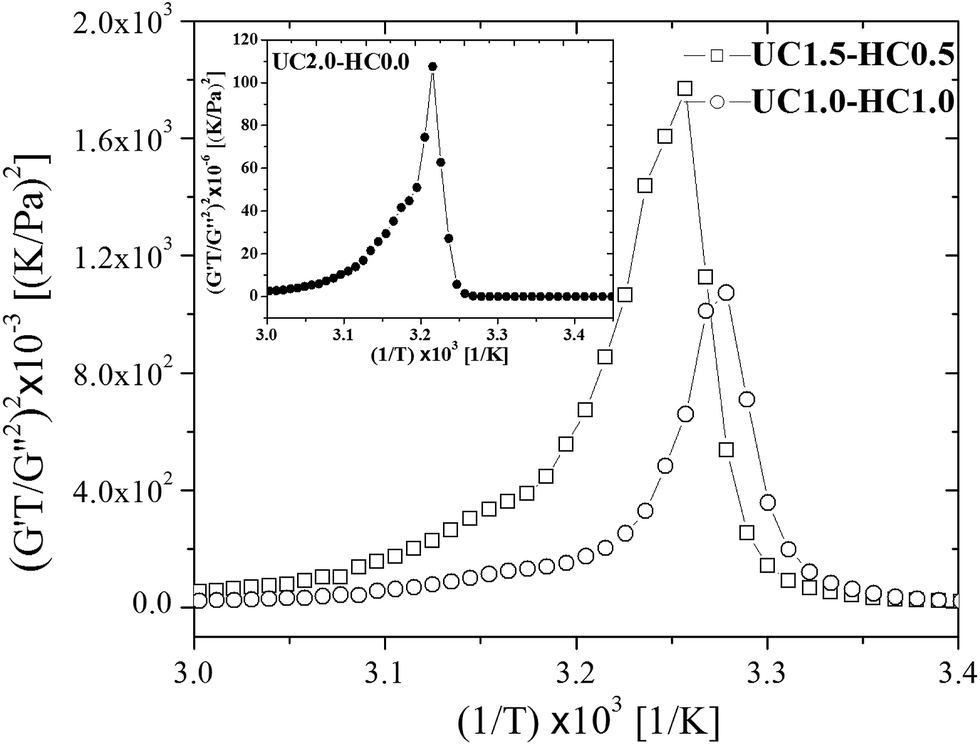 | ||
| Fig. 5 Determination of the gelation temperature based on Fredrickson–Larson theory for UC1.5–HC0.5/GP, and UC1.0–HC1.0/GP samples in comparison with UC2.0–HC0.0/GP system shown in the inset. | ||
In this context it is instructive to recall that the kinship between macroscopic phase separation and gelation is not a new finding, but it has been recognized in a number of studies42–46 on thermoreversible gels. It has been argued that thermoreversible gelation and phase separation are phenomena that are closely correlated to each other. In view of this, we can speculate that as the amount of HC-chains in the solution increases the phase separation with be shifted to lower temperatures and as a consequence the gel temperature should show the same tendency. It has been reported46 from a rheological study on thermoreversible gelling aqueous systems of ethyl(hydroxyethyl)cellulose that phase separation and gelation follow each other and that the gel temperature is lower than the corresponding cloud point temperature. It was argued45 from a Monte Carlo simulation model that due to the coexistence of phase separation and gelation, the cross-linking may arrest the phase-separation process.
3.2. Analysis of turbidity and SALS results
Turbidity measurements were carried out to characterize mesoscopic structural changes of the species developed during sol–gel transition. As can be seen in Fig. 6a and b, it is obvious that the scattered intensity increases with increasing temperature. The overall turbidity changes are reminiscent of the heat-induced evolution of storage modulus, which substantiate similar stages of the gelation process as discussed above. A salient feature in Fig. 6 is the strong upturn of the turbidity in the vicinity of the gel temperatures, indicating the formation of large-scale associations in the systems. At lower temperatures (i.e., the first stage of gelation), the turbidity remains almost constant in the case of UC2.0–HC0.0/GP, whereas a gradual increase is observed for UC2.0–HC0.0/GP. The reason for this is that the growth of the hydrophobic microdomains commences at lower temperatures in the presence of HC chains; this leads to a lower gel-temperature. In the third region, the observed turbidity trend can be associated with the tendency of the system to minimize its interfacial area among microdomains,39 and the late stage gel-stiffening in the systems.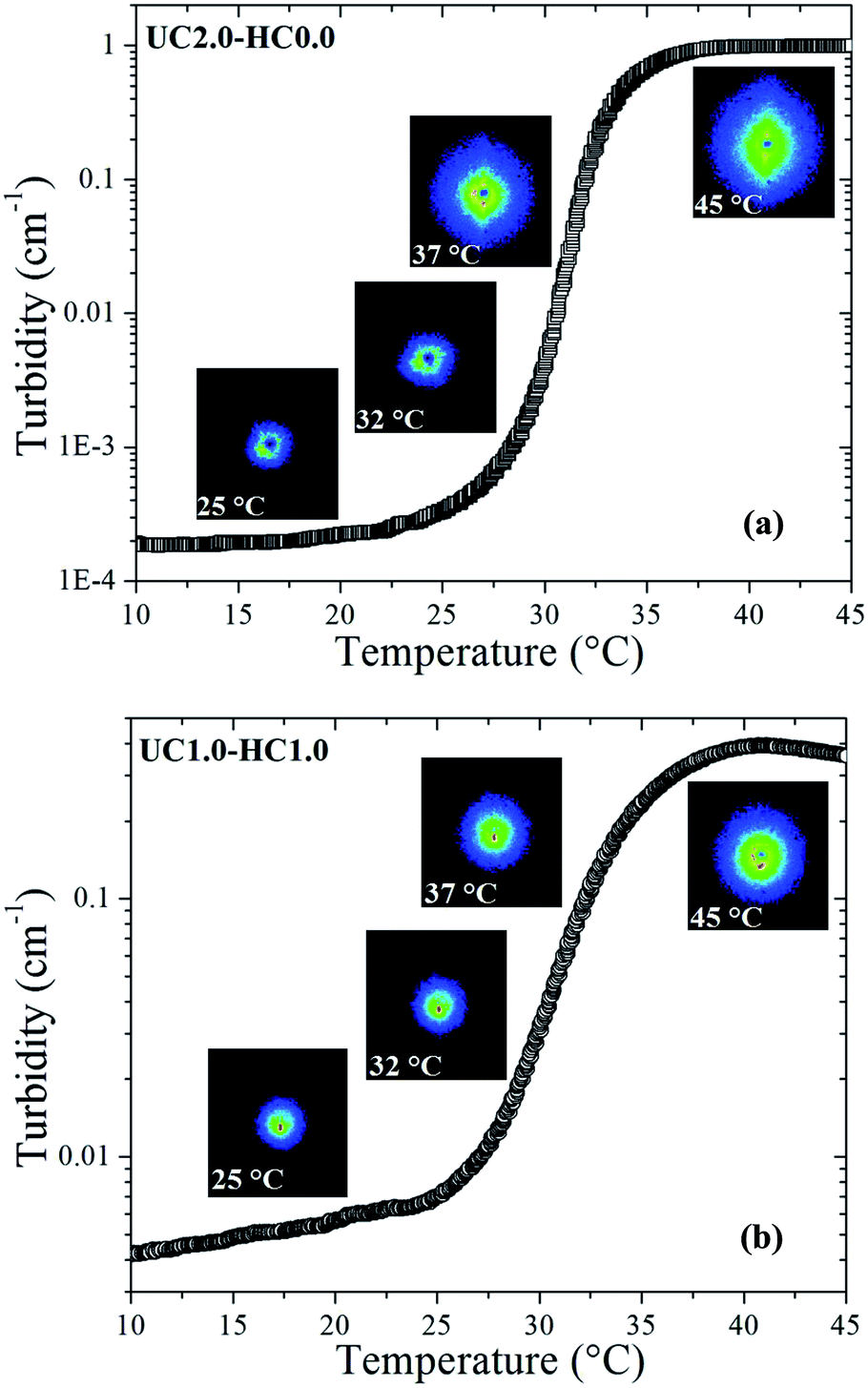 | ||
| Fig. 6 The evolution of the gel networks studied by turbidimetry method during heating from 10 to 45 °C for UC2.0–HC0.0/GP compared to UC1.0–HC1.0/GP. Inset images display SALS 2D patterns at the indicated temperatures. | ||
To gain insight into possible temperature-induced morphological alterations of the gel networks on global dimensional scales, small angle light scattering measurements in the quiescent state were conducted and monitored during the heating course. Typical SALS patterns for the UC2.0–HC0.0/GP and UC1.0–HC1.0/GP systems are depicted in Fig. 6a and b at the indicated temperatures. The obtained patterns support the surmise that large associative domains are formed. As seen, the 2D scattered intensity profiles become stronger with increasing temperature, which corroborates the evolution of the multi-chain complexes.47 Weak intensity patterns are observed at temperatures lower than the gel point (25 and 32 °C for UC2.0–HC0.0/GP, and 25 °C for UC1.0–HC1.0/GP), but augmented intensity patterns are found at elevated temperatures. Patterns of the UC2.0–HC0.0/GP system in the higher temperature range exhibit anisotropic scattered intensity profiles with elliptical scattering patterns. However, in the case of UC1.0–HC1.0/GP, the scattered intensity profiles illustrate more isotropic pattern. This indicates that lower amplitude concentration fluctuations are developed in the presence of HC chains. In other words, the SALS patterns reveal how the structure of the associative complexes can be modulated by contribution of the HC chains. It should be noticed that the obtained scattering patterns are consistent with the above discussed results on the fractal dimension, as well as the strength of the studied thermogelling systems (cf.Fig. 4).
3.3. Gelation time measurements
The gel time is considered as one of the most important features for in situ gel-forming systems. To determine the gelation time of the formulations, time-sweep rheological measurements were performed. Time evolution of the storage and loss modulus at 37 °C for UC2.0–HC0.0/GP and UC1.0–HC1.0/GP systems are depicted in Fig. 7a. In the case of UC1.0–HC1.0/GP, G′ starts to increase at 60 s and this is sooner compared to the UC2.0–HC0.0/GP formulation. It can be argued that the aggregation and percolation of the hydrophobic domains have been expedited in the presence of HC chains, which is consistent with the lower percolation temperature obtained from the temperature sweep rheological results. The observed changes in the time evolution of the storage modulus can be interpreted in accordance with the proposed gelation mechanism.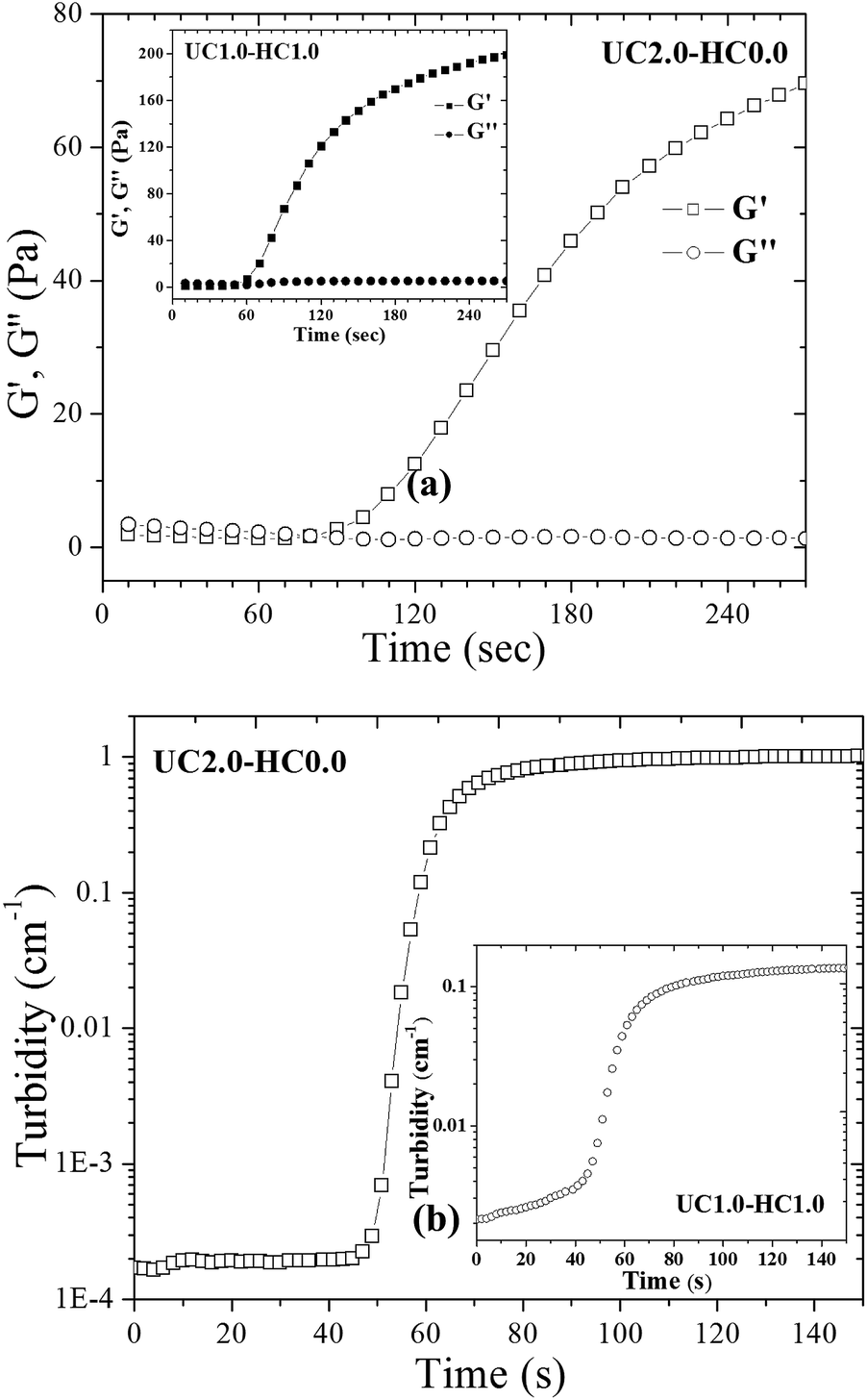 | ||
| Fig. 7 (a) Time dependency of storage (G′) and loss (G′′) modulus at 37 °C for UC2.0–HC0.0/GP in comparison with UC1.0–HC1.0/GP system displayed in the inset. (b) Turbidity changes with time at constant temperature of 37 °C for UC2.0–HC0.0/GP compared to UC1.0–HC1.0/GP system displayed in the inset. | ||
Turbidity behavior of the gelling systems over time was also studied at 37 °C. As is evident from Fig. 7, the turbidity changes of both systems (UC2.0–HC0.0/GP and UC1.0–HC1.0/GP) are similar, and the turbidity exhibits three stages, which are reminiscent of the corresponding three gelling stages of the systems. However, the initial stage in the presence of HC chains shows a continuous increase, indicating the effective role of the hydrophobic moieties on the formation of the multi-chain domains (cf. the inset of Fig. 7b). The features observed here, again emphasize the interference between incipient phase separation and gelation.48
4. Conclusions
In summary, we presented novel in situ gel-forming biopolymer formulations containing β-glycerophosphate disodium salt with tunable gel characteristics. It was demonstrated that incorporation of hydrophobically modified chitosan chains (N-dodecyl chitosan, HC) not only makes the gel point to be shifted to lower temperatures, but also significantly improves the gel strength. In-depth, this rheological investigation showed that the HC chains facilitate heat induced hydrophobic associations followed by percolation of hydrophobic domains and the development of physical gel networks with higher fractal dimensions than the corresponding networks without added HC-chains. The Fredrickson–Larson theory was proposed as an alternative robust and less tedious method (compared to the classic Winter–Chambon theory) to accurately determine the gel point of the studied thermogelling systems. The morphological alterations of the temperature-induced gels by SALS measurements disclosed that a lower amplitude of the concentration fluctuations developed in the presence of HC chains. Time-sweep rheological experiments, as well as turbidity measurements demonstrated that formulations comprising of HC chains undergo faster sol–gel transition. The present findings suggest that the developed chitosan-based hydrogels may serve as promising fine-tunable matrices for various biomedical applications.Acknowledgements
Authors would like to gratefully thank Dr A.-L. Kjøniksen and Dr A. Maleki for helpful scientific discussions and technical assistance. Dr H. Salimi Kenari, Dr M. M. Hasani-Sadrabadi, M. Aliaghaie and R. Nasseri are also thankfully acknowledged for their valuable contributions. E. Dashtimoghadam also highly appreciates Iran's Ministry of Science, Research and Technology for awarding the scholarship.Notes and references
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- C. M. Kirschner and K. S. Anseth, Acta Mater., 2013, 61, 931–944 CrossRef CAS PubMed.
- S. Chaterji, I. K. Kwon and K. Park, Prog. Polym. Sci., 2007, 32, 1083–1122 CrossRef CAS PubMed.
- A. Bajpai, S. K. Shukla, S. Bhanu and S. Kankane, Prog. Polym. Sci., 2008, 33, 1088–1118 CrossRef CAS.
- J. D. Kretlow, L. Klouda and A. G. Mikos, Adv. Drug Delivery Rev., 2007, 59, 263–273 CrossRef CAS PubMed.
- S. R. Van Tomme, G. Storm and W. E. Hennink, Int. J. Pharm., 2008, 355, 1–18 CrossRef CAS PubMed.
- M. H. Park, M. K. Joo, B. G. Choi and B. Jeong, Acc. Chem. Res., 2011, 45, 424–433 CrossRef PubMed.
- Y. Li, J. Rodrigues and H. Tomas, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- D. Y. Ko, U. P. Shinde, B. Yeon and B. Jeong, Prog. Polym. Sci., 2013, 38, 672–701 CrossRef CAS.
- J. Mano, G. Silva, H. S. Azevedo, P. Malafaya, R. Sousa, S. Silva, L. Boesel, J. M. Oliveira, T. Santos and A. Marques, J. R. Soc., Interface, 2007, 4, 999–1030 CrossRef CAS PubMed.
- S. Van Vlierberghe, P. Dubruel and E. Schacht, Biomacromolecules, 2011, 12, 1387–1408 CrossRef CAS PubMed.
- P. B. Malafaya, G. A. Silva and R. L. Reis, Adv. Drug Delivery Rev., 2007, 59, 207–233 CrossRef CAS PubMed.
- T. Coviello, P. Matricardi, C. Marianecci and F. Alhaique, J. Controlled Release, 2007, 119, 5–24 CrossRef CAS PubMed.
- N. Goodarzi, R. Varshochian, G. Kamalinia, F. Atyabi and R. Dinarvand, Carbohydr. Polym., 2013, 92, 1280–1293 CrossRef CAS PubMed.
- H. T. Ta, C. R. Dass and D. E. Dunstan, J. Controlled Release, 2008, 126, 205–216 CrossRef CAS PubMed.
- N. Bhattarai, J. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 83–99 CrossRef CAS PubMed.
- F. S. Majedi, M. M. Hasani-Sadrabadi, J. J. VanDersarl, N. Mokarram, S. Hojjati-Emami, E. Dashtimoghadam, S. Bonakdar, M. A. Shokrgozar, A. Bertsch and P. Renaud, Adv. Funct. Mater., 2014, 24, 432–441 CrossRef CAS.
- H. Sashiwa and S.-I. Aiba, Prog. Polym. Sci., 2004, 29, 887–908 CrossRef CAS.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- A. Chenite, C. Chaput, D. Wang, C. Combes, M. Buschmann, C. Hoemann, J. Leroux, B. Atkinson, F. Binette and A. Selmani, Biomaterials, 2000, 21, 2155–2161 CrossRef CAS PubMed.
- A. Chenite, M. Buschmann, D. Wang, C. Chaput and N. Kandani, Carbohydr. Polym., 2001, 46, 39–47 CrossRef CAS.
- J. Berger, M. Reist, A. Chenite, O. Felt-Baeyens, J. Mayer and R. Gurny, Int. J. Pharm., 2005, 288, 17–25 CrossRef CAS PubMed.
- M. P. Lutolf, P. M. Gilbert and H. M. Blau, Nature, 2009, 462, 433–441 CrossRef CAS PubMed.
- J. J. Rice, M. M. Martino, L. De Laporte, F. Tortelli, P. S. Briquez and J. A. Hubbell, Adv. Healthcare Mater., 2013, 2, 57–71 CrossRef CAS PubMed.
- A.-L. Kjøniksen, C. Iversen, B. Nyström, T. Nakken and O. Palmgren, Macromolecules, 1998, 31, 8142–8148 CrossRef.
- B. Nyström, A.-L. Kjøniksen and C. Iversen, Adv. Colloid Interface Sci., 1999, 79, 81–103 CrossRef.
- E. Dashtimoghadam, H. Mirzadeh, F. A. Taromi and B. Nyström, Polymer, 2013, 54, 4972–4979 CrossRef CAS.
- F. S. Majedi, M. M. Hasani-Sadrabadi, S. H. Emami, M. A. Shokrgozar, J. J. VanDersarl, E. Dashtimoghadam, A. Bertsch and P. Renaud, Lab Chip, 2013, 13, 204–207 RSC.
- A.-L. Kjøniksen, A. Laukkanen, C. Galant, K. D. Knudsen, H. Tenhu and B. Nyström, Macromolecules, 2005, 38, 948–960 CrossRef.
- K. Zhu, H. Jin, A.-L. Kjøniksen and B. Nyström, J. Phys. Chem. B, 2007, 111, 10862–10870 CrossRef CAS PubMed.
- H. H. Winter and F. Chambon, J. Rheol., 1986, 30, 367 CrossRef CAS.
- D. F. Hodgson and E. J. Amis, J. Non-Cryst. Solids, 1991, 131, 913–920 CrossRef.
- H. S. Kenari, M. Imani, E. Dashtimoghadam, A. Maleki, B. Nyström and A. Nodehi, Polymer, 2013, 54, 2999–3007 CrossRef CAS.
- M. Lavertu, D. Filion and M. D. Buschmann, Biomacromolecules, 2008, 9, 640–650 CrossRef CAS PubMed.
- Z. E. Meybodi, M. Imani and M. Atai, Carbohydr. Polym., 2013, 92, 1792–1798 CrossRef PubMed.
- H. Winter, Polym. Eng. Sci., 1987, 27, 1698–1702 CAS.
- M. Muthukumar, Macromolecules, 1989, 22, 4656–4658 CrossRef CAS.
- M. Aliaghaie, H. Mirzadeh, E. Dashtimoghadam and S. Taranejoo, Soft Matter, 2012, 8, 7128–7137 RSC.
- G. H. Fredrickson and R. Larson, J. Chem. Phys., 1987, 86, 1553 CrossRef CAS.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- F. Tanaka and W. H. Stockmayer, Macromolecules, 1994, 27, 3943–3954 CrossRef CAS.
- F. Tanaka, Adv. Colloid Interface Sci., 1996, 63, 23–40 CrossRef CAS.
- F. Tanaka and M. Ishida, Macromolecules, 1996, 29, 7571–7580 CrossRef CAS.
- Y. Liu and R. Pandey, J. Chem. Phys., 1996, 105, 825–836 CrossRef CAS.
- A.-L. Kjøniksen, B. Nyström and B. Lindman, Macromolecules, 1998, 31, 1852–1858 CrossRef.
- Z. Liu, A. Maleki, K. Zhu, A.-L. Kjøniksen and B. Nyström, J. Phys. Chem. B, 2008, 112, 1082–1089 CrossRef CAS PubMed.
- F. Tanaka, Polymer Physics: Applications to Molecular Association and Thermoreversible Gelation, Cambridge University Press, 2011 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |