DOI:
10.1039/C4RA04629C
(Review Article)
RSC Adv., 2014,
4, 35808-35817
Carbon electrodeposition in molten salts: electrode reactions and applications
Received
16th May 2014
, Accepted 4th August 2014
First published on 5th August 2014
Abstract
The chemistry of carbon deposition in molten salts has been studied by a number of researchers with quite different interpretations of the reactions involved. This paper therefore reviews and discusses available literature on the cathodic and anodic reaction mechanisms of carbon electrodeposition in molten carbonate salts, particularly considering new voltammetric studies. The effects of process variables (e.g. salt composition, temperature, voltage, electrode material etc.) on the electrochemical process are also addressed. Description of the properties of the deposited carbon and how these properties transfer to potential applications in energy storage and electricity production (carbon re-oxidation) are discussed.
 Happiness V. Ijije | Happiness V. Ijije received her BEng and MSc degrees from the University of Nottingham in 2010 and 2011 (Malaysia and United Kingdom campuses respectively). She is currently studying for her PhD under the supervision of George Z. Chen. Her research focuses on the electrochemical conversion of CO2 to carbon in molten salts and includes both physical and electrochemical characterizations of the produced carbon. She has first-authored 10 research publications in journals and conferences, and won the Postgraduate Endowed Award from the University of Nottingham in March 2014. |
 Richard C. Lawrence | Richard C. Lawrence completed his MEng and PhD in Chemical Engineering at the University of Nottingham in July 2010 and October 2013, respectively. His PhD research was supervised by Prof. George Z. Chen on the subject of electro-deposition of carbon from molten carbonate salts under carbon dioxide atmospheres, focusing on elucidating the mechanisms of electro-deposition and electro-re-oxidation. Dr Lawrence currently works as a Technical Support Advisor for an effluent treatment plant in the UK civil nuclear industry. |
 George Z. Chen | George Z. Chen (CChem, FRSC, FIMMM, FRSA) is professor of electrochemical technologies in the University of Nottingham. His current research focuses on electrochemical and liquid salts innovations for materials, energy and environment. He obtained a teaching diploma in chemistry from Jiujiang Teacher Training College in 1981 and the MSc degree in physical chemistry from Fujian Normal University in 1984. Under the supervision of Prof. John W. Albery (FRS), he received the PhD degree from the University of London and a Diploma of Imperial College in 1992. Prof. Chen is author/co-author of over 160 peer reviewed articles and 300 conference/seminar presentations. |
1. Introduction
Two significant challenges facing humanity at present are the provision of sustainable and affordable energy, and the avoidance of catastrophic global climate change. Both of these challenges stem from our over-reliance on fossil fuels as the principal energy source. The use of fossil fuels is not sustainable due to their finite reserves, and the combustion of such fuels also leads to the production of greenhouse gasses (e.g. CO2, N2O etc.). In order to reduce CO2 emissions from anthropogenic sources, different carbon abatement technologies (e.g. fuel switching) are currently being considered, developed or employed. The use of biomass to partly meet the world energy demand is one example of fuel switching. Renewable energy technologies (e.g. wind, solar, tidal, biomass and hydropower) have received universal acceptance as clean energy sources but the development is slow, largely due to the significant cost, unsecured availability, intermittency and geopolitical issues. The reliance on fossil fuel as a main energy source is therefore expected to continue for several decades to come. One technique that has been proposed to compromise between the pros and cons of fossil fuels is carbon capture and storage (CCS), in which CO2 is captured, compressed and stored away from the atmosphere for protracted periods of time in geological formations. Alternative to storage, it would be highly desirable to convert the captured CO2 to useful products such as hydrocarbons, methanol and other chemicals.1–3 Electrochemical reduction of CO2 in aqueous solutions is a proven technology and requires the use of appropriate electrolyte, electrode material, catalyst and operating conditions.4–6 An alternative method of converting CO2 to a useful product is to reduce it to solid carbon via electrolysis in molten alkali metal carbonates. In the 1960s, researchers established that carbon could be electro-deposited from molten salts containing carbonate and lithium ions (CO32− and Li+).7–9 More recently, a number of authors have suggested that this process could be utilised for the indirect conversion of CO2 to carbon10–15 through electro-reduction of CO32− ions in the molten salt electrolyte to solid carbon and oxide ions (reaction (1)).7,10,16,17 The oxide ions produced from the reaction can react with CO2 present in the atmosphere above the molten salt to regenerate the CO32− ions (reaction (2)).10| | |
CO32− + 4e− → C + 3O2−
| (1) |
This paper discusses carbon electro-deposition in molten salts. Particular attention is paid to cyclic voltammetry (CV which may also stand for cyclic voltammogram in the following text, depending on the context) carried out on various electrode materials and the different reaction mechanisms involved. The effects of process variables such as the molten salt composition,7 temperature and electrolysis voltage,14,15,17 and gas atmospheres7,15 on the electrolytic process are all considered. A practical application of the process is to convert the deposited carbon back to produce electricity via re-oxidation. Electrical energy used to deposit the carbon is stored as chemical energy in the carbon deposit itself; this energy can be converted back to electricity when the deposit is electrochemically re-oxidised. The uses of carbon in energy storage applications such as supercapacitors are well established and play vital roles in the energy industry.18–20 Utilization of the deposited carbon to fabricate supercapacitors for energy storage application is also discussed.13
2. Mechanisms of the reduction of CO2 to carbon
2.1. Direct reduction of CO2 to carbon
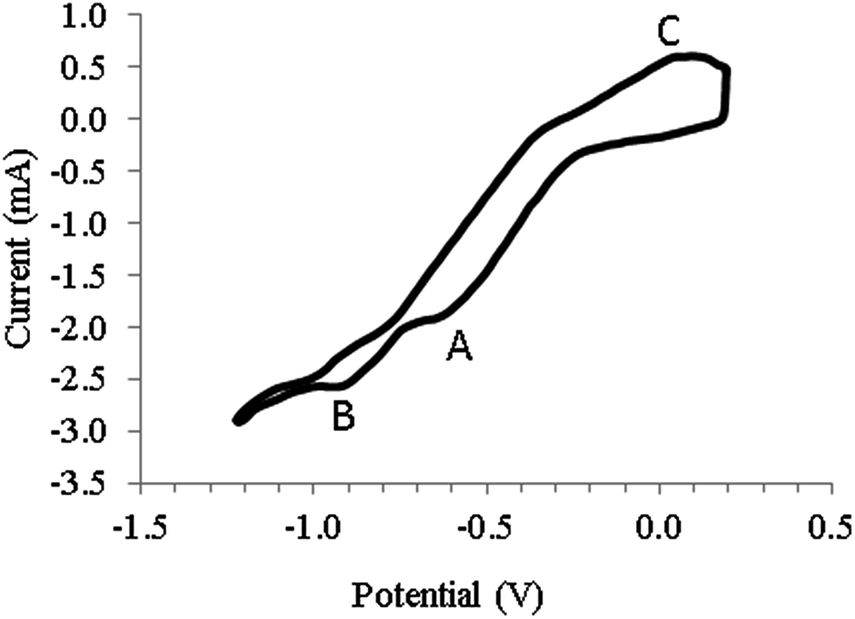
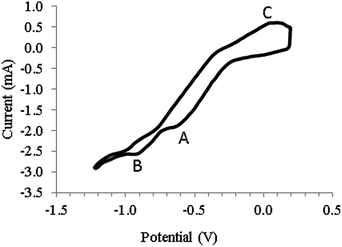
In the direct electrochemical mechanism, CO2 dissolved in a molten salt electrolyte is reduced on the negative electrode (cathode) to elemental carbon.21 Direct electrolysis of CO2 into elemental carbon and oxygen (reaction (3)) was achieved at a pressure of 0.203 MPa.9 Recently a three step electrochemical–chemical–electrochemical CO2 reduction mechanism (reaction (4)–(6)) was proposed.22–25 In this mechanism, CO2 besides being reduced further acts as an acceptor of oxide ions (O2−) and as such CO32− ions are generated. The overall reaction (7) is therefore the reduction of CO2 to carbon and CO32− ions. Evidence for this mechanism was obtained from CVs recorded in the molten mixture of NaCl–KCl (molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 750 °C under a CO2 pressure of 1.0 MPa. Fig. 1 shows the CV. Reversing the scan at the first cathodic peak A resulted in no anodic peak, but when the scan was reversed at peak B at a potential of around −0.9 V vs. Pt, anodic peak C was observed. Carrying out electrolysis in the constant potential mode at the second cathodic peak potential resulted in carbon deposition at the electrode surface. Cathodic peak A was therefore attributed to the quasi reversible reduction of CO2 to a CO22− radical (first stage of the proposed mechanism, reaction (4)) and peak B as the third electro-reduction stage where CO is reduced to carbon and oxide ions (reaction (6)). Direct reduction of CO2 in NaCl–KCl was confirmed by the formation of carbon deposit on the electrode surface at potentials more negative than that peak B. One possible limitation of the study was the failure of the study to mention the possible presence of oxides in the chloride molten salt due to its hygroscopic nature. These oxides could absorb CO2 leading to the formation of CO32− ions which could be electro-reduced to indirectly produce carbon.
:1) at 750 °C under a CO2 pressure of 1.0 MPa. Fig. 1 shows the CV. Reversing the scan at the first cathodic peak A resulted in no anodic peak, but when the scan was reversed at peak B at a potential of around −0.9 V vs. Pt, anodic peak C was observed. Carrying out electrolysis in the constant potential mode at the second cathodic peak potential resulted in carbon deposition at the electrode surface. Cathodic peak A was therefore attributed to the quasi reversible reduction of CO2 to a CO22− radical (first stage of the proposed mechanism, reaction (4)) and peak B as the third electro-reduction stage where CO is reduced to carbon and oxide ions (reaction (6)). Direct reduction of CO2 in NaCl–KCl was confirmed by the formation of carbon deposit on the electrode surface at potentials more negative than that peak B. One possible limitation of the study was the failure of the study to mention the possible presence of oxides in the chloride molten salt due to its hygroscopic nature. These oxides could absorb CO2 leading to the formation of CO32− ions which could be electro-reduced to indirectly produce carbon.
 |
| | Fig. 1 Cyclic voltammogram obtained at a gold working electrode using a NaCl–KCl melt (molar ratio of 1:1) at a temperature of 750 °C under a carbon dioxide pressure of 1.0 MPa. Counter electrode: glassy carbon or platinum crucible; reference electrode: platinum quasi-reference electrode; scan rate: 0.1 V s−1 (This figure is re-plotted from ref. 24.). | |
First step: quasi reversible reduction of CO2 to a CO22− radical.
Second step: chemical formation of CO.
Third step: irreversible electro-reduction of CO to carbon.
Overall reaction:
| | |
3CO2 + 4e− → C + 2CO32−
| (7) |
2.2. Indirect reduction of CO2 in molten carbonates
Indirect reduction of CO2 to carbon in molten alkali or alkaline earth metal carbonates is possible as a result of the equilibrium reaction given in reaction (8) (where M represents any of the corresponding metal). Reduction of CO32− ions to carbon can occur via three different reaction routes. In the first, a direct electrochemical reduction of the CO32− ion proceeds via reaction (1),7,10,16,17,26 occurring either in the presence or absence of CO2 and involving the transfer of four electrons.7,16 Secondly, the CO32− ion can also be reduced via a two-step process involving the hypothetical CO22− ion (reaction (9) and (10)).27 This CO22− ion is thought to have been formed by the combination of a CO molecule and a O2− ion.28 Thirdly, CO32− ions can also be reduced indirectly via the prior reduction of alkali metal ions to the metal (reactions (11) and (12)).29 Linear sweep voltammetry performed on a platinum electrode in molten LiF–NaF–KF–K2CO3 (molar ratio: 45.1:11.2:40.8:2.9) at 500 °C showed the absence of a CO32− ion reduction peak before the cathodic limit, suggesting CO32− ions were not directly reduced.29 It was therefore postulated that CO32− ions are not electrochemically reduced but are rather reduced in a chemical reaction following the formation of the alkali metal.| | |
MxCO3 ⇌ MxO + CO2 (x = 1, 2)
| (8) |
| | |
CO32− + 2e− → CO22− + O2−
| (9) |
| | |
CO22− + 2e− → C + 2O2−
| (10) |
| | |
2xM + MxCO3 → C + 3MxO
| (12) |
Of all three reaction routes, most authors seem to agree that the reduction of CO32− ions occurs via reaction (1) and the corresponding products are carbon and oxide ions.7,10,17,30,31 The O2− ions generated during the reduction process can react with CO2 present in the atmosphere above the molten salt to regenerate more CO32− ions according to reaction (2).10,16 CO2 can therefore be indirectly converted to solid carbon via molten carbonate electrolysis. During electrolysis, the reduction of CO32− ions to CO via reaction (13) is also possible,32 however the principal anodic reaction is the oxidation of CO32− ions to CO2 and O2 via reaction (14).8,17,23,26 Besides the production of O2 at the anode via reaction (14), O2− ions produced at the cathode can migrate towards the anode where they can be oxidised to O2 according to reaction (15). Results obtained via inline gas chromatography of the gases given off at the anode during electrolysis in Li2CO3–Na2CO3–K2CO3 (molar ratio of 43.5:31.5:25.0) at 500 °C under Ar atmosphere showed that both O2 and CO2 were present during the electrolysis but the ratio of CO2/O2 increases with increasing the current density, implying that reaction (14) dominated at higher current densities and at more positive potentials.32 Vigorous bubbling at a Pt anode was also reported during electrolysis in LiF–NaF–Na2CO3 at 700 °C.11
| | |
CO32− + 2e− → CO + 2O2−
| (13) |
| | |
2CO32− → 2CO2 + O2 + 4e−
| (14) |
3. Electrolysis variables
3.1. Molten salt and gas compositions
The molten salt composition has been reported to affect both cathodic and anodic processes. Electrodeposition of carbon from CO32− ions under atmospheric CO2 reportedly only occurred when Li+ ions were present.7 No carbon was deposited during electrolysis in the mixtures of Na2CO3 and K2CO3.7,33 Electrochemical deposition and re-oxidation of solid carbon was reported in molten CaCl2–CaCO3–LiCl–KCl (molar ratio of 0.30:0.17:0.43:0.10) and Li2CO3–K2CO3(62:38) at temperatures between 500 and 850 °C under Ar or CO2 atmospheres.34 The present authors confirmed the absence of carbon deposition at an electrolysis voltage of 4 V in some lithium-free, carbonate-only electrolytes: Na2CO3, K2CO3 and Na2CO3–K2CO3. These findings support the conclusion that the presence of lithium ions is necessary in order for carbon electrodeposition to occur in carbonate-only electrolytes. Thermodynamic calculations can provide answers as to why Li2CO3 reduction to carbon is more favourable than Na2CO3 or K2CO3 under certain conditions. The relevant thermodynamic data for Li2CO3, Na2CO3 and K2CO3 at different temperatures are given in Table 1. It can be seen that the potential required to deposit carbon, E°C, is more positive than that for Li+ ion reduction to Li metal, E°M, in molten Li2CO3, suggesting a preference for carbon deposition. However, in molten Na2CO3 or K2CO3, E°C is comparable or more negative than E°M, which means that reduction of the Na+ or K+ ion is either competitive or preferred to carbon deposition. Note that at temperatures below 700 °C, E°C in molten Na2CO3 is slightly more positive than E°M, although no carbon deposit could be obtained. This may be attributed to kinetics, e.g. the overpotential for carbon deposition is too high. Similar kinetics may apply in molten K2CO3.
Table 1 Standard potentials vs. CO32−/CO2–O2 for the conversion of metal carbonate to pure metal, carbon and carbon monoxide, for Li, Na and K carbonates at different temperatures
| T (°C) |
Metal |
E°M (V) |
E°C (V) |
E°CO (V) |
| Values of E°M and E°C at 450–700 °C were taken from ref. 11, whilst the other values were calculated using data from ref. 35. |
| 450 |
Li |
−3.12a |
−1.78a |
−2.15 |
| Na |
−2.71a |
−2.61a |
−2.60 |
| K |
−2.77a |
−3.15a |
−3.98 |
| 540 |
Li |
−3.01a |
−1.71a |
−2.01 |
| Na |
−2.59a |
−2.53a |
−3.11 |
| K |
−2.65a |
−3.06a |
−3.81 |
| 620 |
Li |
−2.91a |
−1.65a |
−1.89 |
| Na |
−2.49a |
−2.47a |
−2.98 |
| K |
−2.54a |
−2.98a |
−3.66 |
| 700 |
Li |
−2.82a |
−1.59a |
−1.79 |
| Na |
−2.39a |
−2.40a |
−2.86 |
| K |
−2.43a |
−2.90a |
−3.52 |
| 900 |
Li |
−2.50 |
−1.48 |
−1.53 |
| Na |
−2.14 |
−2.26 |
−2.59 |
| K |
−2.19 |
−2.73 |
−3.20 |
Carbon deposition reportedly occurred under N2, CO2 and Ar atmospheres. Current efficiency for carbon deposition in Li2CO3–K2CO3–Na2CO3 at −2.6 V and 600 °C under Ar and CO2 atmospheres was shown to be 97 and 67% respectively.7 The low current efficiency obtained under CO2 was attributed to the reaction of carbon with CO2 according to reaction (16).7
Carbon was also deposited under pure N2 and mixtures of N2 and CO2.15 Under the mixed CO2–N2 atmosphere, carbon deposition became quicker with increasing the CO2 partial pressure.15 Utilisation of CO2 not only limits the molten salt decomposition, but also assists the regeneration of the CO32− ions and thus carbon deposition.
3.2. Temperature and electrode potential
The formation of CO as reported becomes increasingly favourable at higher temperatures (Table 1) and the current efficiency for carbon deposition decreases with the increase in temperature due to reaction (16).32 On the other hand, it is plausible that the drop in current efficiency with temperature could also be a consequence of the production of CO according to reaction (16). Current efficiency was reported to increase when the electrode potential became more negative.7 Efficiencies in excess of 100% were reported at more negative potentials,30 and are likely due to the metal, oxide and carbonate impurities being not excluded during evaluation.
Apart from the oxidation of CO32− ions being dominant at higher current densities, there is very little known on the effect of temperature and electrode potential on anodic reactions. Besides the oxidation of CO32− ions, anodic oxidation of the cathodic product, mostly the O2− ion, is also plausible and varies with the temperature and the electrode potential.
4. Voltammetric studies on different working electrodes
Voltammetry of glassy carbon electrodes showed cathodic and anodic limits, corresponding to the reduction and oxidation of CO32− ions respectively via reactions (1) and (14).17,26,30
Re-oxidation peaks on the CVs from various studies were mostly attributed to the oxidation of the deposited carbon. Carbon deposition was also reported on a gold electrode7,8,16 in Li2CO3–Na2CO3–K2CO3, but another study could not find carbon deposit even though the Au electrode and molten salt used were similar.30 There are also different results on the deposition of carbon on a graphite electrode as carbon deposition in molten Li2CO3 under Ar contradicts reports of the absence of carbon on graphite in Li2CO3–Na2CO3–K2CO3 at temperatures of 500–800 °C under CO2.36,37 Between the two studies, the differences in electrolyte and atmospheres are unlikely to be the cause for the contradictory observations. Possibly the manner in which cathodic polarisation was applied was responsible. Carbon deposition was reported on a Ni electrode.12,17,26,29,34,37,38 Reduction peaks on CVs at −2.0 and −2.4 V vs. Ag in eutectic Li2CO3–Na2CO3–K2CO3 at 450 °C were attributed to the reduction of CO32− ions to carbon and the deposition of alkali metal respectively. The two oxidation peaks observed were assigned to the electrochemical dissolution of the deposited alkali metals and carbon.13 In another study on a Ni electrode under similar conditions to that stated earlier, the cathodic limit (−1.75 V vs. CO2–O2) corresponded to the reduction of CO32− ions to carbon while the anodic limit was assigned to the oxidation of Ni according to reaction (17). The intermediate peaks on the CVs were assigned to presence of nickel oxide.30 Carbon was also deposited on W,11 Ag,38 Mo,36 Al,10 Cu16,30 and glassy carbon26,30 electrodes.
| | |
2Li+ + 2CO32− + 10e− → 6O2− + Li2C2
| (18) |
| | |
PtO32− + 4e− → Pt + 3O2−
| (19) |
| | |
C + 2O2− → CO2 + 4e−
| (20) |
| | |
C + 3O2− ⇌ CO32− + 4e−
| (20a) |
| | |
C + 2CO32− → 3CO2 + 4e−
| (21) |
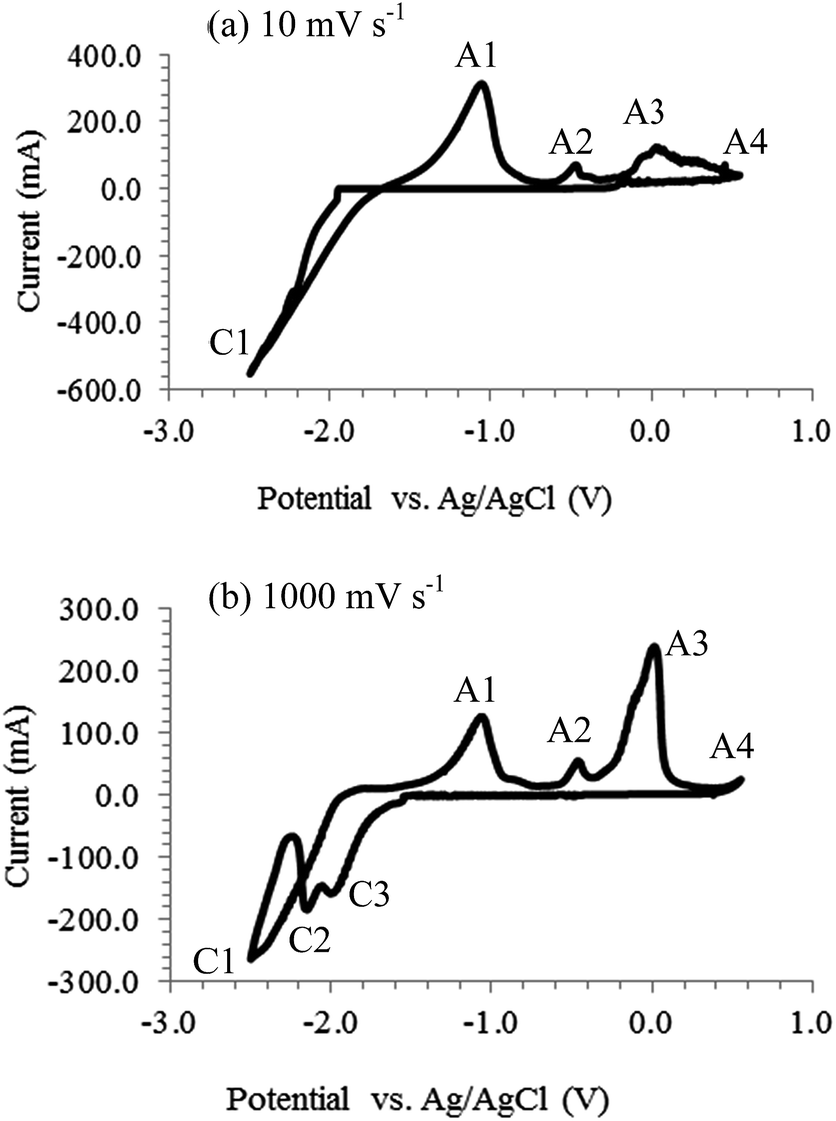
CVs were also recorded on Pt electrodes and carbon deposition was reported.12,30,34,39,40 The CVs in Fig. 2 were obtained by the current authors in molten Li2CO3–Na2CO3 (mole ratio: 52:48). The alumina reference electrode used was described elsewhere.41 Pt was inert in the short duration and specified scan rate of the experiment as these were confirmed by the absence of any reduction peaks before the cathodic limit (−1.7 V). Absence of any cathodic peak before the cathodic limit in Fig. 2a is consistent with that reported before in the same molten salt.40 The cathodic loop observed on the CVs likely represents the formation of a new phase (electrodeposited carbon) on the surface of the working electrode.42
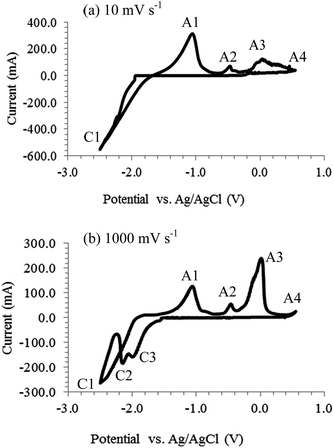 |
| | Fig. 2 Cyclic voltammograms of a 0.25 mm Pt wire electrode in the molten eutectic mixture of Li2CO3–Na2CO3 (mole ratio: 52:48) at 600 °C with a negative potential limit of −2.5 V at (a) 100 mV s−1 and (b) 1000 mV s−1. Atmosphere: CO2; working electrode (WE) immersion depth: ca. 0.9 cm; counter electrode (CE): 6 mm diameter stainless steel rod; reference electrode (RE): alumina membrane Ag/AgCl.41 | |
Carbon was deposited in this study at potentials beyond the cathodic limit. On increasing the potential scan rate, two small cathodic peaks (C2 and C3) became visible as shown in Fig. 2b. Occurrence of these peaks at higher scan rates suggests the corresponding reactions to be kinetically controlled. Based on the thermodynamic potentials given in Table 1, it is likely that carbon monoxide formation via reaction (13) occurred beyond the cathodic limit. Formation of Li2C2 through reaction (18) from the electro-reduction of Li2CO3 cannot be ruled out either and as the standard potential for reaction (18) at 600 °C is −1.92 V which lies within the potential range. The presence of Li2C2 was also mentioned17 and detected in unwashed deposited carbon samples.32,43 Alkali metal production from the reduction of alkali metal ions via reaction (11) is possible as well, since the overpotential for the deposition of alkali metal onto the Pt surface would probably be lower than that for carbon deposition. The formation of Li2PtO3 when Pt was immersed in carbonate-based molten salts was reported44 and as such reaction (19) may be considered.30
Regardless of the specific reactions responsible for cathodic peaks C2 and C3 in Fig. 2b, the fact that they only fully appeared at very high scan rates is evidence that several competing reactions took place at the cathodic limit, including electro-deposition of carbon. Peaks A1 to A3 observed on the CVs during the reverse (anodic) scan should correspond to the oxidation of products from CO32− reduction. While peak A1 is likely reaction (20) or (20a), peak A2 may be attributed to the reverse of (13) or (18), and peak A3 to (21) according to their potentials. However, when the deposited carbon was placed in water, often no bubbling occurred, indicating absence of alkali metals or carbides. Peak A4 appears at the anodic end and must be (14). More discussion on anodic oxidation of the deposited carbon is given below.
CVs were recorded on stainless steel electrodes (Grade 304) in mixed Li2CO3–Na2CO3 (molar ratio: 60:40) and Li2CO3–K2CO3 (62:38) at 650 °C.45 The atmosphere used was humidified anode and cathode gases in molten carbonate fuel cells. Carbon was not intentionally produced during the study, although it was recognised that carbon could be formed and the anodic peak at −1.5 V was attributed to carbon oxidation via reaction (20).45 No carbon was obtained on a stainless steel electrode in molten Li2CO3–Na2CO3–K2CO3 at 600–700 °C.46 This was probably due to the fact that low current densities were employed. Carbon deposition was reported on a stainless steel electrode in Li2CO3–K2CO3 at 540–700 °C.15 The current authors confirmed the deposition of carbon on both stainless and mild steel electrodes at electrolysis voltages of 2.5–5.0 V in molten Li2CO3, Li2CO3–Na2CO3 (mole ratio: 52:48) and Li2CO3–Na2CO3–K2CO3 (mole ratio: 43.5:31.5:25.0).
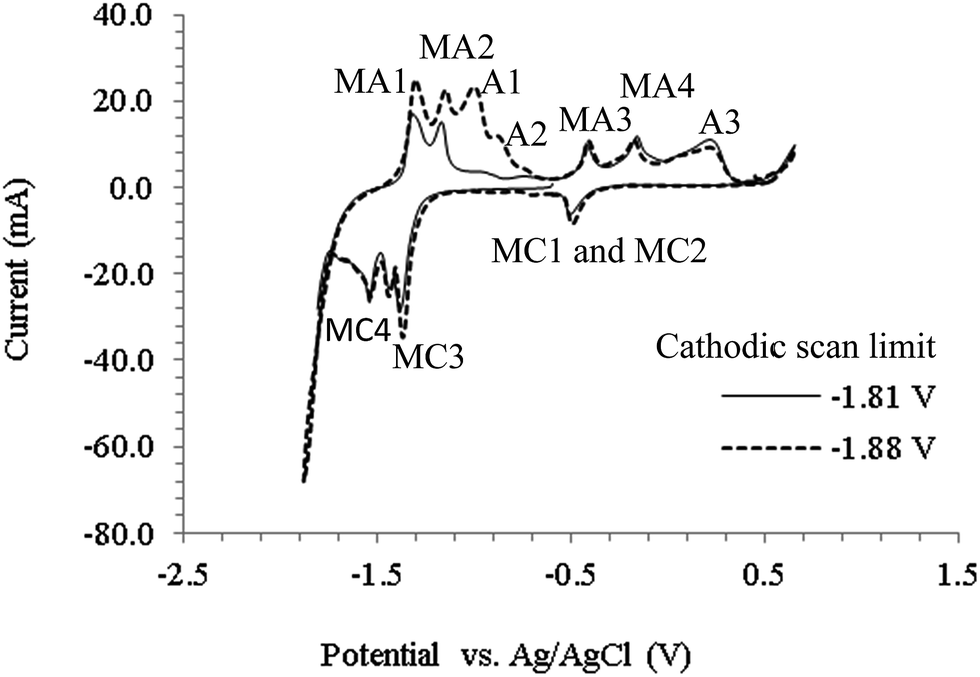
Fig. 3 shows CVs obtained on a 0.25 mm stainless steel electrode in mixed Li2CO3–Na2CO3 (molar ratio of 52:48) at 627 °C. The cathodic limit (−1.71 V) which corresponds to the deposition of carbon is similar to that observed on the Pt working electrode (Fig. 2). Stainless steel comprises of mainly Fe, Ni and Cr and is not as inert in molten carbonate salts as Pt, which explains the various cathodic peaks (marked with M) observed before the cathodic limit (which are not shown in Fig. 2). Based on the findings of this work and those in literature,45,47 it seems likely that these cathodic peaks may be associated with transition metals in stainless steel. As a result, no attempt to identify the specific metal related reactions was made here. Considering the high molten salt temperatures, it is also likely that other competing reactions occurred, such as reduction of alkali metal ion and formation of CO. While research is still ongoing to identify the oxidation peaks, it is likely that the peak A1 corresponds to the re-oxidation of the deposited carbon. By making the potential limit more negative (−1.88 V), the height of peak A1 increased suggesting more carbon was deposited and re-oxidized. Carbon deposition continued to occur at potentials beyond the cathodic limit in all the molten salts studied.
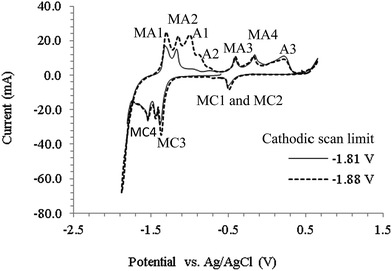 |
| | Fig. 3 Cyclic voltammograms obtained using a stainless steel wire working electrode in a molten Li2CO3–Na2CO3 electrolyte (molar ratio of 52:48) at 627 °C, at a scan rate of 100 mV s−1, for negative potential limits of −1.81 V and −1.88 V. Atmosphere: CO2; WE diameter: 0.25 mm; WE immersion depth: ca. 1.5 cm; CE: 6 mm diameter stainless steel rod; RE: alumina membrane Ag/AgCl. | |
5. Anode material
The choice of anode (counter electrode in cyclic voltammetry) is critical as it must be able to withstand high temperatures, have adequate conductivity and stability, be inert and not undergo any form of chemical dissolution in the molten salt. Unlike the cathode (or working electrode) on which the carbon deposits, the anode surface is always exposed to the molten salts. There is limited literature on the suitability of various anode materials during the electrolysis of molten carbonate salts to produce carbon. Anode materials (Ni, Fe, Cu, Pt, Ir, SnO2 and Ni10Cu11Fe alloy) were compared in studies carried out in a eutectic mixture of Li2CO3–Na2CO3–K2CO3 at 500 °C.13 Dissolution of the Fe and Ni electrodes occurred, a layer of CuO was observed on the surface of a Cu electrode, but no obvious change in diameter of the electrode was noted. Corrosion pits formed at the surface of the Ni10Cu11Fe alloy after 2 h of electrolysis and the SnO2 (Stannex E) anode reportedly did not exhibit any change even after 500 h of electrolysis.13 The conditions (e.g. voltage, temperature, electrolysis mode, i.e. continuous or batch) at which this long time electrolysis was carried out however was not mentioned. SnO2 (Stannex ELR) was studied by the current authors for its suitability in molten Li2CO3–K2CO3 and was found to be quite stable, but very susceptible to cracking due to thermal shocks. Compared to stainless steel, the higher resistivity of SnO2 resulted in voltage losses and as such, the quantity of carbon obtained was less. On the other hand, the interaction of SnO2 with the molten salt was limited, which agrees with the finding that the purity of the carbon products is higher compared to those where stainless steel anode was used. The suitability of stainless steel as the anode material was also investigated by the present authors. Anode passivation dominated at higher temperatures while dissolution of the anode occurred at lower temperatures. At low temperatures, the dissolution of stainless steel also increased with electrolysis voltage.
6. Properties and reuse of the deposited carbon
Regardless of the exact conditions used to deposit carbon, a number of authors have ascertained that the structure of such carbon is primarily amorphous.13,17,27,40 The deposited carbon was also found to possess largely sp2 hybridisation with some carbon at the surface possessing sp3 hybridisation.10 Carbon nanoparticle possessing high specific surface areas also were reported.13,14,17 One report claimed a specific surface area reaching 1315 m2 g−1 for carbon powder deposited at 450 °C from molten Li2CO3–Na2CO3–K2CO3 at an applied potential difference of −6.0 V and dried at 600 °C under vacuum.17 Research by the current authors has yielded carbon deposits with an average specific surface area in the range of 200–700 m2 g−1 without any activation. The existence of graphitic domains has also been reported with fringe spacing of approximately 0.34 nm.17,26 Nearly uniform carbon nano-fibres self-arranged into long ropes (10–50 nano-fibres per rope, average rope diameter of 100–500 nm and maximum rope length of 1 μm) have also been reported.31 Electrolytic conditions such as electrode potential, molten salt choice and temperature, all contribute to the properties of the carbon produced. Increasing molten salt temperature leads to large and elongated carbon particles with low specific surface areas, while more negative potentials increase the specific surface area.17 However, low specific surface areas at more negative potentials were also reported.30 Carbon nano-wires, -flakes and -sheets, and cage like structures were observed under different electrolysis conditions.14 Subjecting the carbon to heat treatment under vacuum increased the degree of surface disordering48,49 but had a relatively weak influence on the specific surface area.17 Compounds such as Li2O2 and Li2C2 were detected depending on treatment conditions.30
The present authors studied the effects of molten salt temperature and composition on the properties of the electro-deposited carbon. Typical resultant morphologies of carbon deposited in Li2CO3–Na2CO3 at 4 V in a two electrode cell are shown in Fig. 4. Aggregated and flat flake-like particles were obtained at 550 °C and 700 °C respectively (Fig. 4a and b). Carbon particles obtained at 700 °C were larger in size compared to those at low temperatures. Flake-like morphology observed in Li2CO3–Na2CO3 at 700 °C (Fig. 4b) is similar to those from pure Li2CO3 and this could be a consequence of the similar deposition temperature. Carbon deposited from Li2CO3–K2CO3 (Fig. 4c) had more aggregated quasi-spherical particles indicating effects of molten salt composition on the properties of deposited carbon. Aggregated carbon particles deposited from Li2CO3–K2CO3 at 700 °C was also similar to those obtained from Li2CO3–Na2CO3 at 550 °C (Fig. 4a). TEM investigations revealed flat fibre-like pattern for carbon deposited from Li2CO3–Na2CO3 at 700 °C (Fig. 4e) while those from Li2CO3 (Fig. 4d) comprised of aggregated particles, fibre-like and possibly rolled up sheets. These observations are in agreement with literatures, that the electrolysis variables all have an effect on the structure and properties of the deposited carbon.
 |
| | Fig. 4 SEM and TEM images of carbon deposited at 4 V in (a and b) Li2CO3–Na2CO3 at 550 and 700 °C respectively, (c) Li2CO3–K2CO3 at 700 °C, (d) Li2CO3 at 722 °C and (e) Li2CO3–Na2CO3 at 700 °C; atmosphere: CO2. | |
Raman spectroscopy of electrodeposited carbon showed both broad G and D peaks,10,13,14 similar to those of graphite (G and D at 1600 and 1350 cm−1 respectively) and was considered to be of sp2 hybridisation. FTIR studies showed absorptions at 3450 and 1110 cm−1 assigned to stretching vibrations of –OH and C–C–O respectively.14 XPS results also showed carbon–carbon bonds, single and double carbon–oxygen bonds,10 and carbonyl and hydroxyl bonds.26
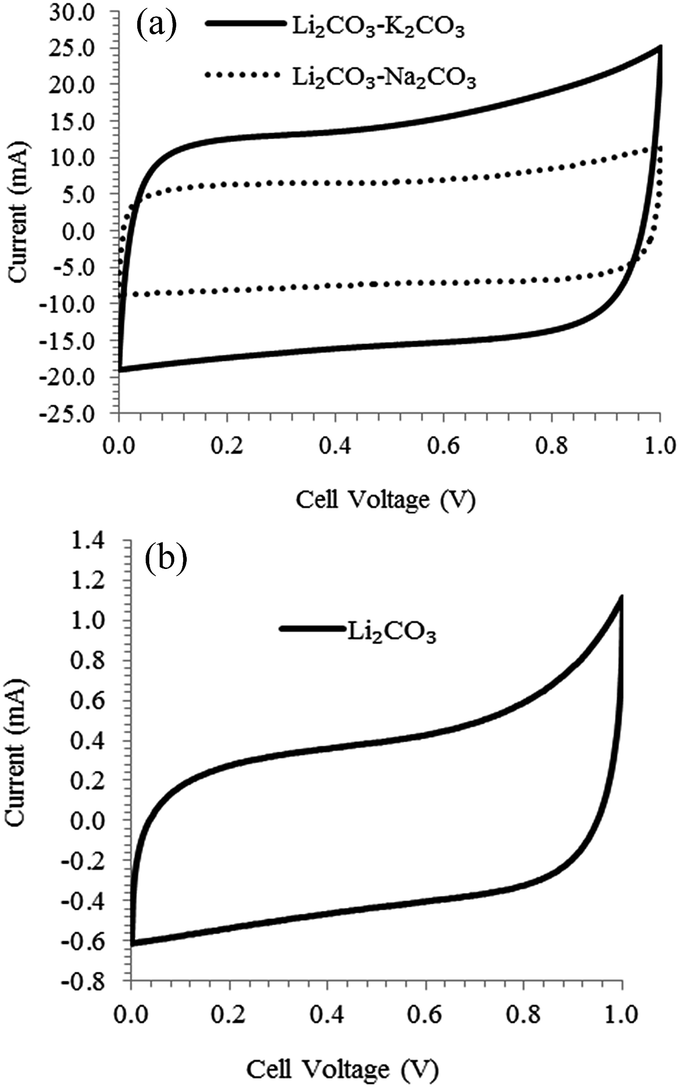
Due to the moderate specific surface areas and the presence of surface functional groups, the electro-deposited carbon has shown promising results in electrochemical energy storage devices such as lithium ion batteries48,49 and supercapacitors.13 In a three electrode cell, specific capacitance reaching 400 F g−1 at a current of 0.2 A g−1 in 1 mol L−1 H2SO4 was reported for carbon deposited at 4 V from Li2CO3–Na2CO3–K2CO3 at 500 °C. (The deposited carbon was mixed with 10 wt% acetylene black and 10 wt% Teflon binder to produce a porous electrode.)13 The current authors used the deposited carbon powders in pellet forms to construct symmetrical two electrode unit cell supercapacitor and the performance in 3 mol L−1 KCl electrolyte was evaluated by CV. The CVs shown in Fig. 5 are almost rectangular, which indicate good reversibility and capacitive behaviour of the carbon electrode. The specific capacitance of carbon deposited in Li2CO3–K2CO3 and Li2CO3–Na2CO3 were 100 and 71 F g−1 with corresponding specific surface area of 535 and 236 m2 g−1 respectively. Considering that the temperature was similar for both molten salts (600 and 632 °C respectively), the change in carbon morphology reflects the effect of the molten salt composition which therefore explains the difference in performance. Carbon deposited in pure Li2CO3 showed very low specific capacitance (2.5 F g−1) compared to the other molten salts which was not unexpected as the specific surface area was only about 20 m2 g−1 (others were above 200 m2 g−1). The large difference in specific capacitance between the 1 mol L−1 H2SO4 electrolyte reported in literature and the 3 mol L−1 KCl used by the present authors is not unexpected. Apart from differences in carbon morphology and electrolyte strength, the size of the solvated ions in the acidic solution is smaller than that in the neutral solution which means that the ions are able to penetrate into the micro pores and as such lead to an increase in the double layer capacitance.50
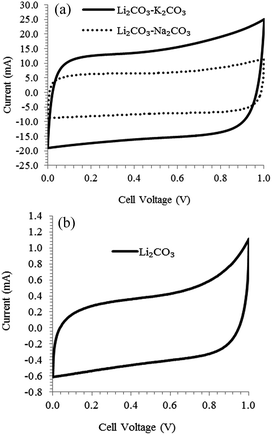 |
| | Fig. 5 CVs of symmetrical unit cells constructed with carbon deposited at a cell voltage of 4 V under CO2 in (a) Li2CO3–K2CO3 (62:38 mol%) at 600 °C and Li2CO3–Na2CO3 (52:48) 632 °C (b) Li2CO3; scan rate: 10 mV s−1; mass of each electrode: 30 mg; electrolyte: 3 mol L−1 KCl; current collector: titanium foil. The carbon material combined with 5 wt% polytetrafluoroethylene (PTFE) was pressed into 13 mm diameter pellets (0.34 mm thick). | |
Besides the moderate specific surface area, low combustion temperatures ranging from 310–450 °C have been reported for carbon deposited from Li2CO3–K2CO3 at various temperatures and voltages.15 This unusual thermal behaviour was attributed to the nano morphology of the electrolytic carbon and the catalytic effects of metal oxide impurities present in the carbon. These factors suggest that the produced carbon could be used as a solid fuel for hi-tech applications. An example could involve using the deposited carbon to make slurries that can be used in internal combustion engines. Although it is possible that deposited carbon can be converted to electricity via the combustion route, it will be more efficient to feed the carbon in a direct carbon fuel cell.
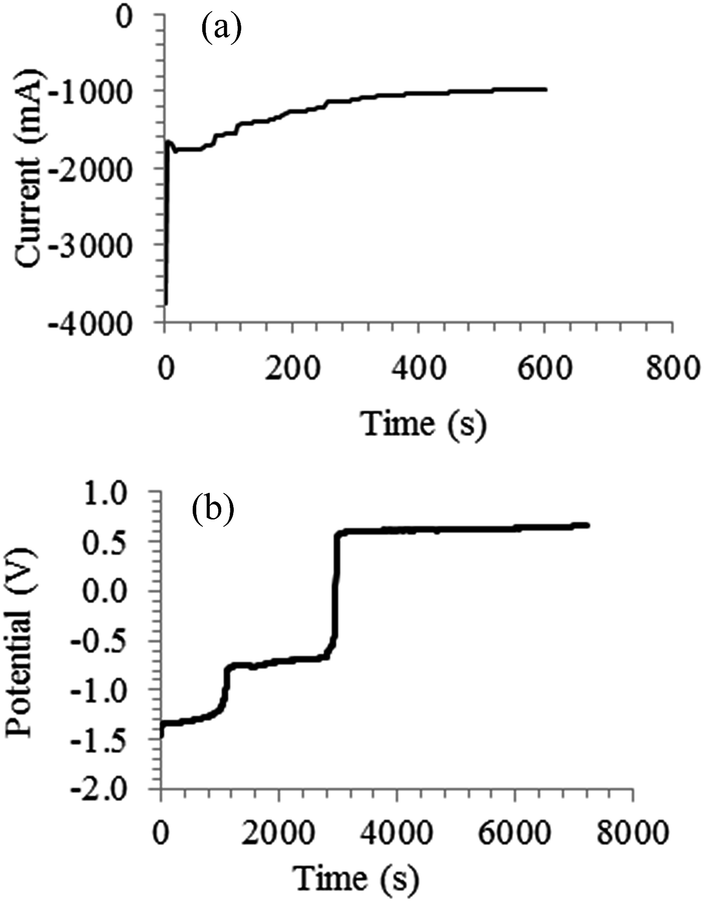
Electrochemical re-oxidation of electro-deposited carbon was only studied in a ternary Li2CO3–Na2CO3–K2CO3 melt at 600 °C, and the parameters varied were the initial quantity of carbon to be re-oxidised and the re-oxidation current.7 The present authors studied the re-oxidation of electro-deposited carbon in molten Li2CO3, Li2CO3–Na2CO3, Li2CO3–K2CO3 and Li2CO3–Na2CO3–K2CO3 with an emphasis on the influence of different molten salts and achievable charge efficiencies. The first step before the re-oxidation of the carbon was actually deposition and a typical chronoamperogram is shown in Fig. 6a. The increase in current with time likely indicates the continuous increase in thickness of the porous carbon deposit. Re-oxidation of the deposited carbon was carried out at 150 mA for 7200 s and a typical potential-time plot for a molten salt studied is given in Fig. 6b. Three potential plateaux were observed. The first plateau at −1.3 V vs. Ag/AgCl is likely the oxidation of carbon according to reactions (20) or (20a). This corresponds well to the first carbon re-oxidation peak A1 in Fig. 1a. The second plateau at −0.7 V should be the oxidation of carbon via reaction (21). The potential is close to that peak A2, but peak A3 may be the true cause. The variations between the plateau and CV peak potentials are expected because the amounts of carbon deposited and the experimental conditions are very different. The third plateau on the other hand matches well with the anodic limit (A4) which is the oxidation of CO32− ions to CO2 and O2. At this plateau there is complete oxidation of the carbon on the electrode surface (no carbon remaining). Carbon was seen on surface of the electrode when the oxidation was stopped at the first and second plateau potentials. The average charge efficiency for the re-oxidation process at the first plateau varied from 15 to 50% while that at the second plateau was in the range of 34 to 95%. The loss of carbon during deposition and re-oxidation of the carbon may explain at least partly those moderate values of charge efficiency obtained. Comparing different molten salts, the charge efficiency for the re-oxidation of carbon in Li2CO3–Na2CO3 is higher than that in Li2CO3–K2CO3 and this may be due to the different properties and structures of the produced carbon as explained previously.
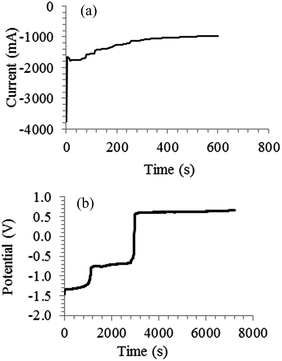 |
| | Fig. 6 (a) Current–time plot recorded at −2.77 V vs. Ag/AgCl during electrolysis in a molten Li2CO3–Na2CO3 (mole ratio: 62:38) at 600 °C on a 5 mm diameter mild steel working electrode; counter electrode: 6 mm diameter stainless steel rod; reference electrode (RE): alumina membrane Ag/AgCl. (b) Potential–time plot recorded during the re-oxidation of carbon deposited in (a); constant current applied: 150 mA; temperature: 600 °C; WE: 5 mm diameter mild steel rod (with carbon deposit still attached); CE: 10 mm diameter graphite rod; alumina membrane Ag/AgCl RE. | |
Aside from simply producing solid carbon, the electrolytic process utilising CO2 can be used to carburise mild steel51 and to produce transition metal carbides.52,53 Carbon films were deposited onto a titanium substrate and enhanced adhesion was observed.43
7. Energy consideration
Energy required for the indirect conversion of CO2 to carbon will be that needed to carry out the electrolysis and heating up of the molten salt.15 The molten salt mixture used for the fixation process should have high chemical absorption capacity for CO2 and the thermal stability of the ionic species in the mixture is also important. The molten salts possess both high thermal and electrochemical stabilities which will lower the cell voltage needed for the electro-reduction. This in turn lowers total energy consumption. Therefore a trade-off between the high thermal and electrochemical stability of different molten carbonate electrolytes is required in order to attain high overall process efficiency. Current efficiencies in the range of 65–100% at electrolysis voltages of 3–5 V for carbon deposition in Li2CO3–K2CO3 (mole ratio: 62:38) at 540–700 °C were reported.15 Higher molten salt temperatures have shown to result in lower current efficiencies7,14,15 and the energy consumption for carbon deposition in eutectic Li2CO3–Na2CO3–K2CO3 at 450 °C ranged from 35–54 kW h kg−1 at 3.5–5.0 V.14 Because most anthropogenic CO2 comes from the use of fossil fuel to generate electricity, it is also essential that the energy used for the molten salt electrolysis process does not originate from fossil fuels. A solar energy driven electrochemical carbon capture process involving indirect conversion of CO2 to both CO and carbon in molten carbonates was proposed.12,54–56 In the electrochemical cell CO2 was captured and converted to solid carbon or CO with 34% to over 50% solar efficiency.12
8. Concluding remarks
Electrochemical conversion of CO2 to carbon can occur directly in molten salts under high CO2 pressures. Indirect reduction of CO2 on the other hand can proceed via the reduction of carbonate ions to carbon and oxide ions in molten carbonate salts. The oxide ions in turn react with the CO2 available from the molten salt atmosphere to regenerate the CO32− ions. From experiments carried out, the presence of Li+ ions is also necessary to deposit carbon in molten carbonate salts as carbon deposition was not obtained in pure molten Na2CO3 or K2CO3 or their mixtures. Cyclic voltammetry on different working electrode materials (Pt and stainless steel) was used to understand the various electrode reactions occurring and these were in agreement with the above conclusions. Process variables such as molten salt composition, electrolysis voltage and temperature were found to influence the properties of the electro-deposited carbon. Applications in which the carbon producing process can be used include the production of carbon electrode material for energy storage devices such as supercapacitors and batteries. Specific capacitance reaching 100 F g−1 in 3 mol L−1 KCl electrolyte was obtained when the deposited carbon was used as the electrode materials. The performance of electro-deposited carbon was quite good considering the moderate specific surface area (535 m2 g−1). Converting the deposited carbon back to electricity via anodic re-oxidation in the same molten salts achieved charge efficiency as high as 95%. With the ease of carbon storage, this high charge efficiency promises seasonal energy storage. That is to use CO2 to collect and store summer solar energy into carbon for winter uses.34
Acknowledgements
This research received financial support from the Royal Society (2007–2009, Brian Mercer Feasibility Award), the EPSRC (2010–2015, Doctor Training Grant, EP/F026412/1, and EP/J000582/1) and the University of Nottingham (2011–2014, Dean of Engineering Scholarships).
Notes and references
- I. Ganesh, Renewable Sustainable Energy Rev., 2014, 31, 221–257 CrossRef CAS PubMed.
- B. Hu, C. Guild and S. L. Suib, J. CO2 Util., 2013, 1, 18–27 CrossRef CAS PubMed.
- S. Saeidi, N. A. S. Amin and M. R. Rahimpour, J. CO2 Util., 2014, 5, 66–81 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, J. CO2 Util., 2013, 3–4, 65–73 CrossRef CAS PubMed.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CAS.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 CAS.
- M. D. Ingram, B. Baron and G. J. Janz, Electrochim. Acta, 1966, 11, 1629–1639 CrossRef CAS.
- Y. K. Delimarskii, O. V. Gordis'kii and V. F. Grishchenko, Dokl. Akad. Nauk SSSR, 1964, 156, 650–651 CAS.
- Y. K. DelimarskiiI, V. I. Shapova, V. A. Vasilenko and V. F. Grishchenko, Otkrytiya, Izobret., Prom. Obraztsy, Tovarnye Znaki, 1975, 52, 176–177 Search PubMed.
- H. Kawamura and Y. Ito, J. Appl. Electrochem., 2000, 30, 571–574 CrossRef CAS.
- G. Chen, Z. Shi, D. Shi, X. Hu, B. Gao, Z. Wang and Y. Yu, Proceedings of 2010 World Non-Grid-Connected Wind Power and Energy Conference, 2010 Search PubMed.
- S. Licht, B. Wang, S. Ghosh, H. Ayub, D. Jiang and J. Ganley, J. Phys. Chem. Lett., 2010, 1, 2363–2368 CAS.
- H. Yin, X. Mao, D. Tang, W. Xiao, L. Xing, H. Zhu, D. Wang and D. R. Sadoway, Energy Environ. Sci., 2013, 6, 1538–1545 CAS.
- D. Tang, H. Yin, X. Mao, W. Xiao and D. H. Wang, Electrochim. Acta, 2013, 114, 567–573 CrossRef CAS PubMed.
- H. V. Ijije, C. Sun and G. Z. Chen, Carbon, 2014, 73, 163–174 CrossRef CAS PubMed.
- L. Massot, P. Chamelot, F. Bouyer and P. Taxill, Electrochim. Acta, 2002, 47, 1949–1957 CrossRef CAS.
- K. Le Van, H. Groult, F. Lantelme, M. Dubois, D. Avignant, A. Tressaud, S. Komaba, N. Kumagai and S. Sigrist, Electrochim. Acta, 2009, 54, 4566–4573 CrossRef CAS PubMed.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS PubMed.
- E. Raymundo-Pinero and F. Beguin, in Activated Carbon Surfaces in Environmental Remediation, ed. T. J. Bandosz, 2006, vol. 7, pp. 293–343 Search PubMed.
- Y. K. Delimarskii, O. V. Gordis'kii and V. F. Grishchenko, Ukr. Khim. Zh., 1965, 31, 32–37 CAS.
- I. A. Novoselova, S. V. Volkov, N. F. Oliinyk and V. I. Shapoval, J. Min. Metall., Sect. B, 2003, 39, 281 CrossRef CAS.
- I. A. Novoselova, N. F. Oliynyk and S. V. Volkov, Hydrogen Materials Science and Chemistry of Carbon Nanomaterials, Springer, 2007, pp. 459–465 Search PubMed.
- I. A. Novoselova, N. F. Oliinyk, A. B. Voronina and S. V. Volkov, Z. Naturforsch., A: Phys. Sci., 2008, 63, 467–474 CAS.
- I. A. Novoselova, N. F. Oliinyk, S. V. Volkov, A. A. Konchits, I. B. Yanchuk, V. S. Yefanov, S. P. Kolesnik and M. V. Karpets, Phys. E, 2008, 40, 2231–2237 CrossRef CAS PubMed.
- B. Kaplan, H. Groult, A. Barhoun, F. Lantelme, T. Nakajima, V. Gupta, S. Komaba and N. Kumagai, J. Electrochem. Soc., 2002, 149, D72–D78 CrossRef CAS PubMed.
- Y. Ito, T. Shimada and H. Kawamura, Proc. – Electrochem. Soc., 1992, 16, 574–585 CAS.
- A. Borucka, J. Electrochem. Soc., 1977, 124, 972–976 CrossRef CAS PubMed.
- M. L. Deanhardt, K. H. Stern and A. Kende, J. Electrochem. Soc., 1986, 133, 1148–1151 CrossRef CAS PubMed.
- F. Lantelme, B. Kaplan, H. Groult and D. Devilliers, J. Mol. Liq., 1999, 83, 255–269 CrossRef CAS.
- B. Kaplan, Chem. Lett., 2001, 714–715 CrossRef CAS.
- P. K. Lorenz and G. J. Janz, Electrochim. Acta, 1970, 15, 1025–1035 CrossRef CAS.
- Y. K. DelimarskiiI, V. F. Grishchenko, N. K. Tumanova and V. I. Shapoval, Ukr. Khim. Zh., 1970, 36, 136–141 Search PubMed.
- H. V. Ijije, R. C. Lawrence, N. J. Siambun, S. Jeong, D. A. Jewell, D. Hu and G. Z. Chen, Faraday Discuss., 2014 10.1039/c4fd00046c.
- M. W. J. Chase, NIST-JANAF Thermochemical Tables, National Institute of Standards, Gaithersberg, 4th edn, 1998 Search PubMed.
- A. T. Dimitrov, Macedonian Source, 2009, 28, 111–118 CAS.
- V. Kaplan, E. Wachtel, K. Gartsman, Y. Feldman and I. Lubomirsky, J. Electrochem. Soc., 2010, 157, B552–B556 CrossRef CAS PubMed.
- G. J. Janz and A. Conte, Electrochim. Acta, 1964, 9, 1269–1278 CrossRef CAS.
- Y. K. DelimarskiiI, V. I. ShapovalL and V. A. Vasilenko, Elektrokhimiya, 1971, 7, 136–141 Search PubMed.
- H. E. Bartlett and K. E. Johnson, J. Electrochem. Soc., 1967, 114, 457–461 CrossRef CAS PubMed.
- H. Wang, N. J. Siambun, L. Yu and G. Z. Chen, J. Electrochem. Soc., 2012, 159, H740–H746 CrossRef CAS PubMed.
- L. Massot, P. Chamelot, F. Bouyer and P. Taxil, Electrochim. Acta, 2003, 48, 465–471 CrossRef CAS.
- Q. Song, Q. Xu, Y. Wang, X. Shang and Z. Li, Thin Solid Films, 2012, 520, 6856–6863 CrossRef CAS PubMed.
- G. J. Janz, A. Conte and E. Neuenschwander, Corrosion, 1963, 19, 292t–294t CrossRef CAS.
- M. Keijzer, G. Lindbergh, K. Hemmes, P. J. J. M. van der Put, J. Schoonman and J. H. W. de Wit, J. Electrochem. Soc., 1999, 146, 2508–2516 CrossRef CAS PubMed.
- G. J. Janz and A. Conte, Electrochim. Acta, 1964, 9, 1279–1287 CrossRef CAS.
- P. Biedenkopf, M. Spiegel and H. J. Grabke, Electrochim. Acta, 1998, 44, 683–692 CrossRef CAS.
- H. Groult, B. Kaplan, S. Komaba, N. Kumagai, V. Gupta, T. Nakajima and B. Simon, J. Electrochem. Soc., 2003, 150, G67–G75 CrossRef CAS PubMed.
- H. Groult, B. Kaplan, F. Lantelme, S. Komaba, N. Kumagai, H. Yashiro, T. Nakajima, B. Simon and A. Barhoun, Solid State Ionics, 2006, 177, 869–875 CrossRef CAS PubMed.
- P. Sharma and T. S. Bhatti, Energy Convers. Manage., 2010, 51, 2901–2912 CrossRef CAS PubMed.
- N. J. Siambun, H. Mohamed, D. Hu, D. Jewell, Y. K. Beng and G. Z. Chen, J. Electrochem. Soc., 2011, 158, H1117–H1124 CrossRef CAS PubMed.
- K. H. Stern, J. Appl. Electrochem., 1992, 22, 717–721 CrossRef CAS.
- K. H. Stern and D. R. Rolison, J. Electrochem. Soc., 1989, 136, 3760–3767 CrossRef CAS PubMed.
- S. Licht, B. Wang and H. Wu, J. Phys. Chem. C, 2011, 115, 11803–11821 CAS.
- S. Licht, J.Phys. Chem C, 2009, 113, 16283–16292 CAS.
- S. Licht, Adv. Mater., 2011, 23, 5592–5612 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a