
Enhanced turnover rate and enantioselectivity in the asymmetric epoxidation of styrene by new T213G mutants of CYP 119†
Abstract
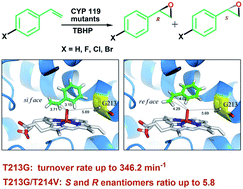
New CYP 119 T213G mutants were constructed and characterized. Introduction of the T213G mutation into the wild-type CYP 119 significantly enhances the turnover rate for the peroxide-dependent styrene epoxidation 4.4-fold to 346.2 min−1, and the double T213G/T214V mutant improves the ratio of the S- and R-enantiomers of the epoxide products 2- fold to 5.8. The molecular modelling and docking results support our initial design and experimental data.
 Please wait while we load your content...
Please wait while we load your content...

