
Validation of the 2,3-dihydroxy-propionyl group in selenium speciation by chemical synthesis and LC-MS analyses†
Abstract
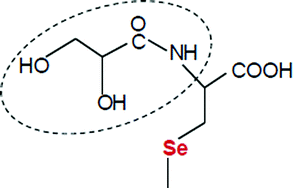
2,3-Dihydroxy-propionyl (2,3-DHP) group is a specific residue detected in selenized yeast that forms numerous stable and highly abundant Se species in several different yeast strains and fermentation batches. The conjugated form of 2,3-DHP-selenocysteine and glutathione is one of the most abundant species that is found in nearly all selenized yeast. In order to overcome the commercial unavailability of this compound, its synthesis was carried out through the active ester formation of pentachlorophenyl glycerate with selenocysteine, followed by the redox conjugation with glutathione. The optimization process of the synthesis was utilized for the production of three other Se-yeast specific compounds, namely, the conjugate of glutathione and selenocysteine, the conjugate of 2,3-DHP-selenocysteine and selenocysteine, and di-N-2,3-dihydroxy-propionyl-selenocysteine. The upstream and clean-up procedures were supported and monitored with HPLC-UV, HPLC-ICP-MS, and HPLC-ESI-QQQMS set-ups, while the identification was performed with HPLC-ESI-QTOFMS. The synthesized 2,3-DHP-selenocysteine/selenocysteine conjugates possessed fragmentation patterns identical to literature data.
 Please wait while we load your content...
Please wait while we load your content...

