 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Semi-crystalline block copolymer bicontinuous nanospheres for thermoresponsive controlled release†
Simon J. Holder*a,
Glen Woodwarda,
Beulah McKenzieab and
Nico A. J. M. Sommerdijkb
aFunctional Materials Group, University of Kent, Canterbury, CT2 8EN, UK. E-mail: S.J.Holder@kent.ac.uk; Tel: +44 1227 823542
bLaboratory of Materials and Interface Chemistry and Soft Matter Cryo-TEM Research Unit, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
First published on 4th June 2014
Abstract
We demonstrate the controlled release of pyrene, as a model hydrophobic molecule, from self-assembled bicontinuous nanospheres formed from an amphiphilic block copolymer. The bicontinuous polymer nanospheres act as efficient nanocarriers and the incorporation of hydrophobic poly(alkyl methacrylate) blocks introduces a temperature responsive component to the hydrophobic core.
Block copolymer self-assembled aggregates have long been studied as nanocarriers for the controlled delivery of encapsulated chemicals, with particular focus on the controlled delivery of pharmaceuticals in biomedical applications. To date most research has focussed on the use of spherical micelles and vesicles (polymersomes) with some studies utilising cylindrical micelles.1,2 We have recently demonstrated the existence of self-assembled bicontinuous polymer nanospheres (BPNs) from amphiphilic block copolymers with low weight fractions of hydrophilic blocks (<25%).3–5 Whilst micelles and vesicles have primarily been studied for the encapsulation and release of lipophilic and hydrophilic compounds respectively, BPNs offer the potential for dual release formulations (e.g. a lipophilic and a hydrophilic pharmaceutical) stemming from the coexistence of the hydrophilic and hydrophobic interior regions. Herein we present the results from our studies into successfully encapsulating a hydrophobic compound (pyrene) into such nanospheres, and the subsequent controlled release of pyrene as a function of temperature.
Thermoresponsive polymers, macromolecules that undergo a physical property change in response to a change in temperature, have been proposed for a number of potential applications. One of the most common being for controlled release from discrete self-assembled aggregates such as micelles and vesicles in biomedical and pharmaceutical uses.1,2 Most of these thermoresponsive systems have been based upon poly(N-isopropylacrylamide) and poly(oligoethylene glycol)6 and other hydrophilic polymers.7 Upon heating above a given temperature (the lower critical solution temperature) such materials become insoluble. A different approach to utilising thermoresponsive polymers has been described using poly(trimethylene carbonate) wherein a melting transition for a hydrophobic polymer results in a profound change in permeability.8,9 Unfortunately polymers for which crystallinity is a property of the main chain have limitations, particularly with respect to manipulation of the melting temperature. A promising approach to the use of semi-crystalline polymers for temperature responsive applications is the incorporation of the crystalline segment as a component of the side-chain; i.e. removed from the backbone.10 By changing the alkyl chain length the melting temperature can be modulated.11 The poly(alkyl (meth)acrylates) are ideal candidates for this approach.12 In this study we demonstrate the potential of this alternative whereby we use poly(octadecyl methacrylate) as semi-crystalline polymer component in a block copolymer bicontinuous nanosphere and demonstrate the release of pyrene across a range of temperatures from 10 to 40 °C.
We prepared poly(ethylene oxide)-block-poly(octadecyl methacrylate) (PEO-b-PODMA) bicontinuous nanospheres in aqueous solution by the slow addition of deionised water to a THF solution of a PEO45-b-PODMA20 copolymer (Mn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900, Mw/Mn = 1.26, by GPC) followed by dialysis against water at 10 °C (ESI†).5 The resulting BPNs displayed a bicontinuous internal structure of interpenetrating PODMA and PEO–water domains with average pore diameters of 20 nm as characterised by cryo-TEM and cryo-ET. The resulting size distribution was monomodal with a z-average diameter of 180 ± 3 nm, a number-average diameter of 127 ± 8 nm and polydispersity index of 0.27 by DLS and 235 ± 16 nm by TEM (Fig. 1a, c and ESI†). BPNs resemble cubosomes generated by molecular amphiphiles which are usually stabilised by PEO based additives.13 Such cubosomes have been proposed and studied for a number of controlled delivery applications.14,15 Whilst our bicontinuous polymeric nanospheres have lower degrees of order than those of their molecular analogues they are easily prepared, their components (PEO biocompatible corona and alkyl chain interiors) are intrinsic to the structure and open to ready modification, and crucially they are kinetically stable. By following identical preparation conditions but using a THF solution of pyrene (Py) in place of pure THF we were able to form bicontinuous nanospheres with encapsulated Py with a monomodal distribution with a z-average diameter of 268 ± 23 nm, a number-average diameter of 141 ± 37 nm, a polydispersity index of 0.27 by DLS (number average) and an average diameter of 222 ± 86 nm by TEM (Fig. 1b, d and ESI†). Py was chosen as a model for the controlled release of hydrophobic compounds owing to its hydrophobic character with very low water solubility (typically in the range 2 × 10−7 to 10 × 10−7 mol dm−3).16,17 Since a dialysis (at 10 °C) was involved in the preparation, it was necessary to demonstrate that a significant amount of Py was still present and retained in the nanospheres. This was demonstrated by analysis of the fluorescent spectra of the Py encapsulated PEO–PODMA solution with that of a Py solution prepared in the absence of the copolymer (Fig. 2). Three aspects of the emission spectra demonstrated encapsulation: (i) a significant difference in emission intensities for otherwise identical quantities of Py, indicative of solubilised Py; (ii) the comparison of the spectra gave I3:I1 ratios (I1 is the 0–0 vibronic band at 372 nm and I3 is the 0–737 band at 383 nm) of 1.03 for the encapsulated Py and 0.67 for the Py in water, indicative of Py in a relatively weakly polar environment for the micellar dispersion, and a highly polar environment in water;5,18,19 (iii) a strong excimer emission (λmax = 471 nm) for the Py in the micellar dispersion; indicative of localised high concentrations of Py.
900, Mw/Mn = 1.26, by GPC) followed by dialysis against water at 10 °C (ESI†).5 The resulting BPNs displayed a bicontinuous internal structure of interpenetrating PODMA and PEO–water domains with average pore diameters of 20 nm as characterised by cryo-TEM and cryo-ET. The resulting size distribution was monomodal with a z-average diameter of 180 ± 3 nm, a number-average diameter of 127 ± 8 nm and polydispersity index of 0.27 by DLS and 235 ± 16 nm by TEM (Fig. 1a, c and ESI†). BPNs resemble cubosomes generated by molecular amphiphiles which are usually stabilised by PEO based additives.13 Such cubosomes have been proposed and studied for a number of controlled delivery applications.14,15 Whilst our bicontinuous polymeric nanospheres have lower degrees of order than those of their molecular analogues they are easily prepared, their components (PEO biocompatible corona and alkyl chain interiors) are intrinsic to the structure and open to ready modification, and crucially they are kinetically stable. By following identical preparation conditions but using a THF solution of pyrene (Py) in place of pure THF we were able to form bicontinuous nanospheres with encapsulated Py with a monomodal distribution with a z-average diameter of 268 ± 23 nm, a number-average diameter of 141 ± 37 nm, a polydispersity index of 0.27 by DLS (number average) and an average diameter of 222 ± 86 nm by TEM (Fig. 1b, d and ESI†). Py was chosen as a model for the controlled release of hydrophobic compounds owing to its hydrophobic character with very low water solubility (typically in the range 2 × 10−7 to 10 × 10−7 mol dm−3).16,17 Since a dialysis (at 10 °C) was involved in the preparation, it was necessary to demonstrate that a significant amount of Py was still present and retained in the nanospheres. This was demonstrated by analysis of the fluorescent spectra of the Py encapsulated PEO–PODMA solution with that of a Py solution prepared in the absence of the copolymer (Fig. 2). Three aspects of the emission spectra demonstrated encapsulation: (i) a significant difference in emission intensities for otherwise identical quantities of Py, indicative of solubilised Py; (ii) the comparison of the spectra gave I3:I1 ratios (I1 is the 0–0 vibronic band at 372 nm and I3 is the 0–737 band at 383 nm) of 1.03 for the encapsulated Py and 0.67 for the Py in water, indicative of Py in a relatively weakly polar environment for the micellar dispersion, and a highly polar environment in water;5,18,19 (iii) a strong excimer emission (λmax = 471 nm) for the Py in the micellar dispersion; indicative of localised high concentrations of Py.
 | ||
| Fig. 1 TEM micrographs of PEO-b-PODMA micelles (a) with no pyrene (no staining), (b) with pyrene encapsulated (negative staining). Cryo-TEM micrographs of bicontinuous nanospheres (c) without pyrene and (d) with pyrene, illustrating internal morphology. | ||
 | ||
| Fig. 2 Normalised fluorescence emission spectra of pyrene in water (red line) and encapsulated in bicontinuous micelle (black line). The inset shows the spectra prior to normalisation. | ||
We previously reported that upon heating from 5 °C to 45 °C a phase transition at 20–25 °C was observed for the bicontinuous components of the PODMA regions of the block copolymer.5 To study the release of Py into the aqueous environment upon heating, samples of the Py–PEO–PODMA dispersion were isolated from pure water (3 dm3) by a dialysis membrane and kept at a set temperature (10, 20, 25, 30 and 40 °C). The relative quantity of Py released into the larger aqueous environment was determined by recording fluorescent spectra of samples at given times over a 6 to 7 hour period (Fig. 3a). A calibration curve of Py in water was used to determine Py concentration variance with time (see ESI†). To confirm that the release of the Py was occurring from the nanospheres the BPN solutions were also analysed by fluorescence spectroscopy after dialysis. In all cases a clear decrease in emission intensity was observed corresponding to loss of pyrene from the nanospheres (Fig. 3b). The release efficiencies were estimated from integration of the curves relative to the undialysed pyrene containing BPN sample (see ESI†). As can be seen (Fig. 3b) a relative maximum in release efficiency (72% release of encapsulated pyrene) was observed at 40 °C. These results also allowed an estimate of the loading capacity and gave a value of 43 μg pyrene per mg of BPN (circa 4% loading). A full study of release efficiency involving infinite sink conditions is needed and is currently underway.
 | ||
| Fig. 3 Release of pyrene from bicontinuous nanospheres formed from PEO45-b-PODMA20, (a) release profiles at various temperatures and (b) photoluminescent spectra of Py nanosphere dispersions before and after dialysis at stated temperatures. | ||
The release profiles for 10 °C, 20 °C and 25 °C are very similar, but for 30–40 °C there is an increasing rate of release of Py (Fig. 3a). The plot of fractional release against time from 10 to 60% release (see ESI†) is linear indicating zero order kinetics and Case-II transport properties (a non-Fickian diffusion release mechanism predominates). Fractional release times were based on extrapolation of the curves in Fig. 3a to constant values resulting from saturation of the water solution by Py which varies with temperature (see ESI†).16 The change in release rates (taken from the slope of the linear fits, ESI†) show the dramatic change from below 25 °C to above 25 °C (Fig. 4a), with fractional release rate constants of 0.0030 s−1 at 25 °C and 0.0051 s−1 at 30 °C and 0.01003 s−1 at 40 °C. This demonstrates the effect of the melting of the hydrophobic PODMA block on Py release and the potential of poly(alkyl methacrylates) in thermoresponsive controlled delivery applications.
 | ||
| Fig. 4 (a) Time taken for indicated percentage release of pyrene from bicontinuous nanospheres at given temperatures. (b) Comparison of fractional release profiles of ‘free’ pyrene (see text) and pyrene encapsulated in bicontinuous nanospheres. | ||
As has become increasingly apparent over the past few years, dynamic equilibrium dialysis conditions are not always suitable for reliably determining the release kinetics of lipophilic compounds from solid nanoparticles and micelles.20–24 To a large part the apparent release kinetics in such set-ups are a result of the partition coefficient between particle and lipophilic compound within the dialysis chamber, any binding to the dialysis membrane (potentially complicated by nanoparticle effects on the membrane itself) and diffusion rates across the dialysis membrane any and all of which can be the limiting factor in the rate of appearance of the lipophile in the receiving chamber. This can complicate data interpretation and can lead to incorrect conclusions about the release kinetics.25 As a control experiment, an identical procedure was followed whereby the release of Py from a dispersion prepared without the PEO–PODMA present was monitored. Since Py is only sparingly water soluble the bulk of the Py remained undissolved. Transfer of the solid Py–water mixture to dialysis tubing and monitoring of the release profiles showed that release of Py into surrounding water was significantly faster than that of the encapsulated Py at 40 °C and at 10 °C (Fig. 4b). However the difference in release rates for ‘free’ and encapsulated pyrene and change in rates with temperature for the encapsulated pyrene indicate a controlled release mechanism.
Despite the reservations currently held about the full validity of dynamic equilibrium dialysis in studying release mechanisms, a number of drug dissolution models were fitted to the fractional release profiles namely the Higuchi, zero order, Weibull and Korsmeyer–Peppas (power law) models.26–31 Of these the Korsmeyer–Peppas model most closely matched the release profiles (between 10 and 60% release) (see ESI†). This model takes the form
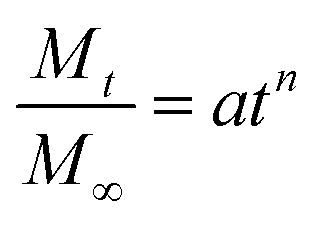
 | ||
| Scheme 1 Illustration of potential regions for pyrene inclusion in BPN. | ||
In summary we have demonstrated that self-assembled bicontinuous nanospheres formed from amphiphilic block copolymers can be used to encapsulate and therefore transport hydrophobic compounds in water. They have been demonstrated to display controlled release and furthermore the release rate can be significantly increased through an increase in temperature.
Finally we note that the melting temperatures of the poly(alkyl(meth)acrylates) are readily altered through the use of different lengths of alkyl chains as substituents and our current work is investigating raising the transition temperatures to greater than 30 °C.
Notes and references
- A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef PubMed.
- U. Kedar, P. Phutane, S. Shidhaye and V. Kadam, Nanomedicine: Nanotechnology, Biology and Medicine, 2010, 6, 714–729 CrossRef CAS PubMed.
- B. E. McKenzie, J. F. de Visser, H. Friedrich, M. J. M. Wirix, P. H. H. Bomans, G. de With, S. J. Holder and N. A. J. M. Sommerdijk, Macromolecules, 2013, 46, 9845–9848 CrossRef CAS.
- B. E. McKenzie, S. J. Holder and N. A. J. M. Sommerdijk, Curr. Opin. Colloid Interface Sci., 2012, 17, 343–349 CrossRef PubMed.
- B. E. McKenzie, F. Nudelman, P. H. Bomans, S. J. Holder and N. A. Sommerdijk, J. Am. Chem. Soc., 2010, 132, 10256–10259 CrossRef CAS PubMed.
- H. Wei, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, Prog. Polym. Sci., 2009, 34, 893–910 CrossRef CAS PubMed.
- S. Hocine and M.-H. Li, Soft Matter, 2013, 9, 5839–5861 RSC.
- C. Sanson, O. Diou, J. Thevenot, E. Ibarboure, A. Soum, A. Brûlet, S. Miraux, E. Thiaudière, S. Tan and A. Brisson, ACS Nano, 2011, 5, 1122–1140 CrossRef CAS PubMed.
- C. Sanson, J. F. Le Meins, C. Schatz, A. Soum and S. Lecommandoux, Soft Matter, 2010, 6, 1722–1730 RSC.
- G. Street, D. Illsley and S. Holder, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1129–1143 CrossRef CAS.
- K. O'Leary and D. R. Paul, Polymer, 2004, 45, 6575–6585 CrossRef PubMed.
- E. Hempel, H. Huth and M. Beiner, Thermochim. Acta, 2003, 403, 105–114 CrossRef CAS.
- P. T. Spicer, Curr. Opin. Colloid Interface Sci., 2005, 10, 274–279 CrossRef CAS PubMed.
- C. Y. Guo, J. Wang, F. L. Cao, R. J. Lee and G. X. Zhai, Drug Discovery Today, 2010, 15, 1032–1040 CrossRef CAS PubMed.
- S. B. Rizwan, B. J. Boyd, T. Rades and S. Hook, Expert Opin. Drug Delivery, 2010, 7, 1133–1144 CrossRef CAS PubMed.
- F. P. Schwarz, J. Chem. Eng. Data, 1977, 22, 273–277 CrossRef CAS.
- T. A. Andersson, K. M. Hartonen and M.-L. Riekkola, J. Chem. Eng. Data, 2005, 50, 1177–1183 CrossRef CAS.
- D. C. Dong and M. A. Winnik, Can. J. Chem., 1984, 62, 2560–2565 CrossRef CAS.
- S. Abraham, T. D. Atvars and R. G. Weiss, J. Phys. Chem. B, 2010, 114, 12221–12233 CrossRef CAS PubMed.
- B. J. Boyd, Int. J. Pharm., 2003, 260, 239–247 CrossRef CAS.
- C. Washington, Int. J. Pharm., 1989, 56, 71–74 CrossRef CAS.
- Y. Zambito, E. Pedreschi and G. Di Colo, Int. J. Pharm., 2012, 434, 28–34 CrossRef CAS PubMed.
- H. Bunjes, J. Pharm. Pharmacol., 2010, 62, 1637–1645 CrossRef CAS PubMed.
- C. Washington, Int. J. Pharm., 1990, 58, 1–12 CrossRef CAS.
- S. Modi and B. D. Anderson, Mol. Pharm., 2013, 10, 3076–3089 CrossRef CAS PubMed.
- P. Costa and J. M. Sousa Lobo, Eur. J. Pharm. Sci., 2001, 13, 123–133 CrossRef CAS.
- T. Higuchi, J. Pharm. Sci., 1963, 52, 1145–1149 CrossRef CAS.
- C. G. Varelas, D. G. Dixon and C. A. Steiner, J. Controlled Release, 1995, 34, 185–192 CrossRef CAS.
- F. Langenbucher, J. Pharm. Pharmacol., 1972, 24, 979–981 CrossRef CAS PubMed.
- W. Weibull, J. Appl. Mech., 1951, 18, 293–297 Search PubMed.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef CAS.
- Y. Fu and W. J. Kao, Expert Opin. Drug Delivery, 2010, 7, 429–444 CrossRef CAS PubMed.
- D. De Kee, Q. Liu and J. Hinestroza, Can. J. Chem. Eng., 2005, 83, 913–929 CrossRef CAS.
- S. C. George and S. Thomas, Prog. Polym. Sci., 2001, 26, 985–1017 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods and techniques; Py in water fluorescence spectra and calibration curve; diffusion modelling fitting plots for PEO–PODMA nanospheres, micelles and pure Py; TEM of PEO–PODMA micelles. See DOI: 10.1039/c4ra04547e |
| This journal is © The Royal Society of Chemistry 2014 |