
The fast C(3P) + CH3OH reaction as an efficient loss process for gas-phase interstellar methanol†
Abstract
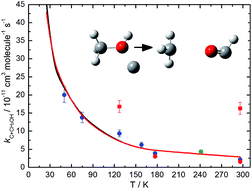
Rate constants for the C(3P) + CH3OH reaction have been measured in a continuous supersonic flow reactor over the range 50 K ≤ T ≤ 296 K. C(3P) was created by the in situ pulsed laser photolysis of CBr4, a multiphoton process which also produced some C(1D), allowing us to investigate simultaneously the low temperature kinetics of the C(1D) + CH3OH reaction. C(1D) atoms were followed by an indirect chemiluminescent tracer method in the presence of excess CH3OH. C(3P) atoms were detected by the same chemiluminescence technique and also by direct vacuum ultra-violet laser induced fluorescence (VUV LIF). Secondary measurements of product H(2S) atom formation have been undertaken allowing absolute H atom yields to be obtained by comparison with a suitable reference reaction. In parallel, statistical calculations have been performed based on ab initio calculations of the complexes, adducts and transition states (TSs) relevant to the title reaction. By comparison with the experimental H atom yields, the preferred reaction pathways could be determined, placing important constraints on the statistical calculations. The experimental and theoretical work are in excellent agreement, predicting a negative temperature dependence of the rate constant increasing from 2.2 × 10−11 cm3 molecule−1 s−1 at 296 K to 20.0 × 10−11 cm3 molecule−1 s−1 at 50 K. CH3 and HCO are found to be the major products under our experimental conditions. As this reaction is not considered in current astrochemical networks, its influence on interstellar methanol abundances is tested using a dense interstellar cloud model.
 Please wait while we load your content...
Please wait while we load your content...

